Pancreatic Cancer
INTRODUCTION
For most patients, pancreatic adenocarcinoma remains highly lethal and is the fourth leading cause of deaths from cancer in the USA. Less than 5% survive 5 years after diagnosis. Surgical resection is the only curative treatment. However, the cure rate with surgery is only 18%–25% and most patients are not surgical candidates. Patients with unresectable disease can have symptoms palliated by chemotherapy and/or radiation therapy. However, these have not significantly impacted 5-year survival. Improved understanding of pancreatic cancer biology continues to provide new therapeutic ideas. Trials are evaluating whether new approaches to earlier diagnosis or improvements in radiation therapy, chemotherapy (including targeted therapy), and/or immunotherapy (e.g., vaccines, therapy aimed at inhibiting negative immunoregulatory proteins) can impact survival.
INCIDENCE AND EPIDEMIOLOGY
• Approximately 45,000 individuals develop pancreatic cancer yearly in the United States with over 38,000 dying from the disease.
• Increases with age, slight male predominance, increased incidence in African Americans, variation in prevalence by world region (higher in western Europe, Scandinavia, the United States, and New Zealand) (1).
• Risk factors for pancreatic adenocarcinoma include (1–3):
Environmental
• Cigarette smoking, history of diabetes mellitus, previous radiation therapy to the pancreas as treatment for other malignancies (such as Hodgkin’s disease, or testicular cancer) and chronic pancreatitis (especially that due to genetic risk factors); increased body mass index and heavy alcohol consumption (but not light consumption) may be risk factors.
Genetic
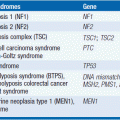
• Mutations in p16; mismatched repair genes (hMSH2 and hMLH1); BRCA1 (rare pancreatic cancers); BRCA2; PALB2 (a BRCA associated protein), STK11/LKB1 (Peutz-Jeghers syndrome); ataxia telangectasia (AT); p53 (Li-Fraumeni syndrome); APC (familial adenomatous polyposis); von Hippel-Lindau (VHL); cationic trypsinogen (PRSS1 gene); and cystic fibrosis transmembrane regulator (CFTR) genes (2–4).
• Families with increased risk of pancreatic cancer without as yet defined genetic abnormalities.
• Overall, approximately 5%–10% of patients with pancreatic cancer will have a first-degree relative who develops pancreatic cancer (2–4).
PATHOLOGY
Normal pancreatic cell types include ductal, acinar, endocrine/neuroendocrine, connective tissue support, endothelial, and lymphocytes. Malignancies can arise from each cell type. In adults, approximately 90% are adenocarcinomas derived from duct cells with approximately two-thirds arising in the head, and one-third being in the body/tail or multicentric (5–7). Other histologic subtypes of ductal origin include pleomorphic carcinomas, giant cell carcinomas, microglandular adenocarcinomas, and cystic neoplasms. Cystic neoplasms comprise a small but increasingly identified subgroup of pancreatic tumors (7). They can be divided into serous cyst adenomas (usually benign) and mucinous cystadenocarcinomas. A higher percentage of these tumors occur in middle-aged women as compared to ductal adenocarcinomas. They appear to be divided into a group that has benign or borderline malignant cells with good prognosis and a group with carcinoma that metastasizes widely and has a prognosis similar to that of other ductal adenocarcinomas. Pancreatic papillary cystic tumors tend to occur in women of reproductive years with relatively better prognosis after surgical excision. There are also noncystic mucin-producing tumors of the pancreas that tend to have a better prognosis after surgical excision. Acinar cell carcinomas make up 1%–2% of pancreatic cancers. Acinar cell tumors occur most commonly in the elderly, but they also occur in younger patients and comprise a higher percentage of tumors seen in children, who have a better prognosis (8). Overall, adult patients with acinar cell carcinomas tend to have a slightly better clinical course than those with ductal adenocarcinomas. Uncommon pancreatic tumors include pancreatic inflammatory tumors and small cell undifferentiated carcinomas. Tumors with mixed histologies including adenosquamous carcinomas and carcinosarcomas can occur and tend to have a poor prognosis. Pancreatoblastomas are rare neoplasms arising from multipotential cells that can differentiate into mesenchymal, endocrine, or acinar cells, which occur primarily in children, although rare cases can occur in adults. They frequently have elevated alpha feto protein levels and are potentially curable when localized. Metastatic pancreaticoblastomas are often responsive to chemotherapy. Other pancreatic tumors found in children include solid pseudopapillary tumors, pancreatic endocrine/neuroendocrine neoplasms, and acinar cell tumors (8).
Pancreatic neuroendocrine cell cancers (PNET) comprise approximately 5%–10% of pancreatic tumors (9). Although associated with longer survival than pancreatic adenocarcinomas, they frequently metastasize. Lymphomas, sarcomas, and other mesenchymal tumors (e.g., teratomas, schwannomas, and neurofibromas) make up only a small proportion of pancreatic cancers (less than 2%). Their biology is similar to that of malignancies of similar histology arising in other areas of the body.
A wide variety of neoplasms can metastasize to the pancreas, including breast, lung, melanoma, renal, gastrointestinal, and other sites.
BIOLOGY OF PANCREATIC ADENOCARCINOMA
Pancreatic adenocarcinomas have frequently invaded locally and/or metastasized by the time initially detected. Local spread is directly to soft tissues and adjacent organs (5, 6). They tend to metastasize widely including lymph nodes, liver, peritoneum, lungs, adrenal glands, and, less commonly, bone and brain.
The biology of pancreatic ductal adenocarcinoma has provided targets for earlier diagnosis, potential therapy, and possible avenues for prevention in the future (3, 4, 10, 11). As is true of most tumors, there is a complex pattern of genetic changes with variation between different tumors. Frequent mutations have been found in proteins involved in cell signaling pathways (especially K-ras [70%–90%]) as well as a number of tumor suppressor genes (especially p53, p16, and DPC4/Smad4). A number of growth factor receptor families, including insulin-like growth factor receptors, the epidermal growth factor receptors (EGFRs), and fibroblast growth factor receptors, are highly expressed in a proportion of pancreatic adenocarcinomas. The life span of cells normally is limited by shortening of telomeric DNA at chromosomal ends. Telomerase (the enzyme important in maintaining telomeric DNA at chromosomal ends) activity is elevated in a high percentage of pancreatic carcinomas.
The potential role of some of the mutations found in familial pancreatic cancer (e.g., BRCA2, PALB2, or mismatched repair genes) in the development of nonhereditary pancreatic cancers is unclear (3, 4, 10, 11). The frequency of these mutations in sporadic cases is low. Tumors that have mutations in mismatched repair genes but not Ras genes are characterized by the appearance of “pushing borders” on histopathology and a better prognosis.
In addition to genetic changes, epigenetic factors, alterations in metabolic processes, suppression of host immune response, hypoxia-induced changes in tumor cells and the microenvironment, and the complexity of the tumor microenvironment all play important roles in development, survival, proliferation, and metastatic spread of pancreatic cancer (10, 11). These all offer potential targets to improve treatment.
PRESENTING SYMPTOMS AND SIGNS
The initial symptoms produced by pancreatic cancer are insidious. Most patients have nonspecific symptoms for several months prior to diagnosis. Tumors in the head of the pancreas sometimes produce obstruction of the bile duct and therefore jaundice at a relatively earlier stage, although most of these tumors are still unresectable. Fatigue, weight loss, anorexia, abdominal pain, back pain, jaundice/light stools/dark urine/pruritus (for head lesions), nausea, vomiting, early satiety, dyspnea, and glucose intolerance are the most common presenting symptoms. Depression is seen in a significant percentage of patients. There is a relatively high incidence of blood clot formation and some patients present with thrombophlebitis. Patients who develop portal or splenic vein obstruction can present with hematemesis (due to varices) or ascites. Ascites may also be due to metastatic disease to the peritoneum.
DIAGNOSTIC WORKUP
The diagnosis should be considered in individuals who present with the above symptoms, especially with several symptoms. A careful history should be obtained including review for the above symptoms, history of cigarette smoking or other risk factors, and a family history. Physical exam should include evaluation for evidence of weight loss, lymph node enlargement (especially in the supraclavicular or periumbilical areas), jaundice, hepatosplenomegaly, ascites, peripheral edema, and evidence of coagulopathy. For most patients, findings on physical exam are nonspecific.
Laboratory tests should include a complete blood count (CBC) and liver function tests, although, in general, laboratory studies are nonspecific and not particularly helpful in making a diagnosis. CA19-9 is the most useful tumor marker and is elevated in 70%–90% of patients with advanced disease. Although not useful as a screening tool due to relative nonspecificity, it is helpful in following a patient’s therapeutic response. Although less frequent, CEA is occasionally elevated in patients who do not have CA19-9 elevations and can be used to follow response to therapy.
Radiological evaluation plays a key role in the diagnosis (12). Computerized tomographic (CT) scans currently are the most commonly used modality for assessing for a pancreatic mass, potential vascular invasion, and determining whether the tumor has metastasized. Alternatively, MRI can be utilized. Pulmonary metastases in the absence of abdominal metastases are relatively uncommon but can occur, and CT imaging of the chest is also important.
Pathology is ultimately required to make a diagnosis. Biopsies of either the pancreas or nodal lesions can be obtained at the time of endoscopic ultrasound (EUS). These theoretically carry less potential risk of peritoneal seeding than percutaneous biopsies. Although ideally a diagnosis can be made preoperatively, for patients with a potentially resectable pancreatic mass, surgery is often necessary in any case even if the initial fine needle biopsy was not diagnostic. For patients with unresectable disease, percutaneous biopsy under radiological guidance of either the pancreas itself or a metastatic lesion is usually obtained.
STAGING AND TREATMENT DECISIONS
The American Joint Commission on Cancer (AJCC) staging system with the TNM format is utilized to group patients into stages I–IV (5
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


