Overview of Megakaryocyte and Platelet Biology
Joel S. Bennett
Platelets are circulating anucleate fragments of bone marrow megakaryocytes that provide primary hemostasis by forming hemostatic plugs at sites of vascular injury; likewise, platelet thrombi are predominantly responsible for the morbidity and mortality of arterial vascular disease. The discovery of platelets as formed elements of the blood, the identification of their central role in hemostasis and thrombosis, and the research that has led to our current understanding of the mechanism and regulation of platelet production and function have been outlined in excellent reviews by Coller1 and Coller and Shattil.2 The chapters in this section of the textbook provide detailed analyses of the current understanding of platelet production and function.
PLATELET PRODUCTION FROM MEGAKARYOCYTES
Megakaryocyte development and the subsequent production and release of platelets are unique phenomena that are discussed in Chapters 23, 24 and 25. Megakaryocytes are relatively rare cells in the bone marrow, constituting <1% of marrow cells, but they are also the largest marrow cells with diameters of up to 40 to 50 mm. They are located in the extravascular compartment of the bone marrow along the albuminal face of the sinusoidal endothelium, frequently in clusters (FIGURE 22.1). Despite their apparent scarcity, sufficient numbers of megakaryocytes are present in the marrow to produce the 1011 platelets per day required to maintain the normal steady-state level of circulating platelets.3 In order to meet this need, megakaryocytes have evolved a unique maturation process in which the diploid DNA of megakaryocyte precursors undergoes repeated cycles of duplication (endomitosis), reaching a modal DNA level of 16N or greater, but without cell division.4 This results in a cell with a single polylobulated nucleus and with an expansive cytoplasm whose fragmentation is the source of circulating platelets. Although the mechanism responsible for endomitosis in megakaryocytes is not known, it is thought to be related to failure to develop and sustain a cleavage furrow during the late stage of mitosis with subsequent absence of cytokinesis. Recent evidence indicates that this is associated with the RUNX1-induced downregulation of the nonmuscle myosin IIB heavy chain,5 a component of the contractile ring that generates the forces necessary for cytokinesis, and with downregulation of the RhoA-specific guanine-nucleotide exchange factors (GEFs) GEF-H1 and ECT2,6 thereby preventing the RhoA signaling required for the formation of the contractile ring.
As discussed in Chapter 24, the conversion of megakaryocyte cytoplasm into platelets has been studied predominantly in vitro using cultured murine megakaryocytes. This process begins with the generation of large pseudopod-like structures from one pole of the megakaryocyte that elongate, in a microtubuledriven process, into long thin branching tubular projections termed proplatelets, which are themselves comprised of tandem arrays of platelet-sized swellings connected by thin cytoplasmic bridges.7 When this process was observed in vivo in mouse skull bone marrow using multiphoton intravital microscopy, megakaryocytes were observed to extend cellular processes, either pseudopodia or immature proplatelets, into bone marrow sinusoids.8 This implies that intravascular shear forces may help to separate proplatelets from the megakaryocytes and that proplatelet morphogenesis into platelets occurs in the peripheral blood.
The primary humoral regulator of megakaryocyte proliferation and differentiation is thrombopoietin (TPO), a 331-amino acid homologue of erythropoietin and a member of the four-helix bundle family of cytokines3 (see Chapter 25). TPO is synthesized predominantly by the liver, but constitutive TPO mRNA expression can be detected in kidney, smooth muscle cells, and other tissues as well. Moreover, TPO mRNA transcription can be induced in bone marrow stromal cells by substantial degrees of thrombocytopenia, and transcription in the liver can be enhanced by the inflammatory response mediator IL-6, perhaps accounting for the reactive thrombocytosis that often accompanies inflammatory states. However, absence of TPO or its receptor (see below) does not preclude platelet production, and platelet counts remain approximately 10% of normal following their genetic elimination in mice. This suggests that other hematopoietic cytokines also support platelet production. Although IL-3, IL-6, and IL-11 stimulate megakaryocyte colony formation in vitro, crossing TPO-receptor null mice with mice null for signaling by these cytokines does not eliminate basal thrombopoiesis. On the other hand, the chemokine SDF-1 synergizes with TPO to support megakaryopoiesis, suggesting that it may support at least a portion of basal platelet production.
The receptor for TPO on megakaryocytes and platelets is c-Mpl, the protooncogene of the murine myeloproliferative leukemia virus and a member of the type 1 hematopoietic cytokine receptor family.3,4 c-Mpl is also present on hematopoietic stem cells that require TPO stimulation for maintenance and expansion. The c-Mpl gene encodes a 610-amino acid protein that resides on cell surfaces as a constitutive homodimer. TPO binding to the homodimer extracellular domain is thought to induce a conformational change in the homodimer that enables autophosphorylation and activation of JAK family kinases (JAK2 and TYK2) associated with its cytoplasmic domains. In addition to phosphorylating the c-Mpl cytoplasmic domains
themselves, thereby providing binding sites for signaling molecules, activated JAK phosphorylates a number of signaling molecules that promote cell survival and proliferation, including members of the signal transducer and activator of transcription (STAT 1, 3, and 5), MAPK, and phosphotidylinositol-3 kinase pathways, as well as proteins that limit cell signaling including the phosphatases SHP1 and SHIP1 and members of the suppressors of cytokine signaling (SOCS) family. There are also a small of number of copies of c-Mpl expressed on the platelet surface (25 to 100 per platelet). Although TPO binding to platelet c-Mpl does not by itself result in platelet activation, it does prime platelets for activation by the classical platelet agonist, perhaps enhancing platelet reactivity in the presence of thrombocytopenia. On the other hand, TPO binding to c-Mpl on platelets and megakaryocytes is thought to play an important role in regulating the concentration of TPO in plasma. TPO adsorbed to the platelet surface is internalized and metabolized. Thus, in the presence of thrombocytopenia, the TPO concentration in plasma increases, stimulating megakaryocyte proliferation; conversely, the TPO concentrations decrease in the presence of thrombocytosis because of an increase in platelet-mediated clearance.9 The low TPO concentrations observed in autoimmune thrombocytopenia has been attributed to increased TPO clearance by the hyperplastic megakaryocytes seen in this disorder.10
themselves, thereby providing binding sites for signaling molecules, activated JAK phosphorylates a number of signaling molecules that promote cell survival and proliferation, including members of the signal transducer and activator of transcription (STAT 1, 3, and 5), MAPK, and phosphotidylinositol-3 kinase pathways, as well as proteins that limit cell signaling including the phosphatases SHP1 and SHIP1 and members of the suppressors of cytokine signaling (SOCS) family. There are also a small of number of copies of c-Mpl expressed on the platelet surface (25 to 100 per platelet). Although TPO binding to platelet c-Mpl does not by itself result in platelet activation, it does prime platelets for activation by the classical platelet agonist, perhaps enhancing platelet reactivity in the presence of thrombocytopenia. On the other hand, TPO binding to c-Mpl on platelets and megakaryocytes is thought to play an important role in regulating the concentration of TPO in plasma. TPO adsorbed to the platelet surface is internalized and metabolized. Thus, in the presence of thrombocytopenia, the TPO concentration in plasma increases, stimulating megakaryocyte proliferation; conversely, the TPO concentrations decrease in the presence of thrombocytosis because of an increase in platelet-mediated clearance.9 The low TPO concentrations observed in autoimmune thrombocytopenia has been attributed to increased TPO clearance by the hyperplastic megakaryocytes seen in this disorder.10
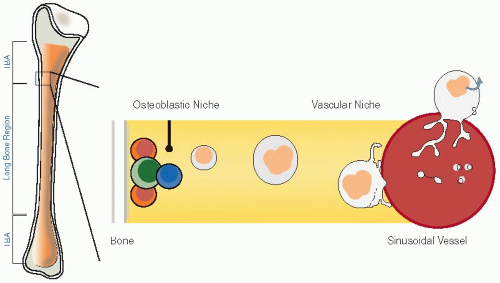 FIGURE 22.1 Diagrammatic representation of megakaryocyte production and platelet production. Hematopoietic stem cells located in the osteoblastic niche next to the endosteal bone surface migrate to the center of the marrow cavity where they undergo successive cycles of endomitosis to produce polyploidy megakaryocytes. Mature megakaryocytes then extend proplatelet elongations into marrow sinusoidal blood vessels where the proplatelets are released and ultimately release individual platelets. (Adapted from Thon JN, Italiano JE. Platelet formation. Semin Hematol 2010;47: 220-226, with permission.) |
ELEMENTS OF PLATELET FUNCTION
Platelet Adhesion
Platelets circulate for 7 to 10 days in a nonreactive state in humans until they are needed for hemostasis. The nonreactive state of circulating platelets is maintained, at least in part, by the inhibitory effect of nitric oxide and prostacyclin secreted by endothelial cells, which increase the platelet content of cyclic AMP (cAMP) and cyclic GMP (cGMP), respectively,11 by the ability of the endothelial cell ecto-ADPase CD39 to metabolize ambient plasma adenosine diphosphate (ADP),12 through the inhibitory effect of platelet proteins such as PECAM-1,13,14 and because the negative surface charge of circulating platelets inhibits their interaction with each other and with erythrocytes, leukocytes, and the vascular endothelium. Further, the intact endothelium presents a barrier separating platelets from adhesive substrates in the subendothelial connective tissue matrix. Disruption of the endothelial barrier by trauma, or by a disease such as atherosclerosis, allows platelets to come in contact with and adhere to the subendothelial matrix.
At the low shear rates present in the venous circulation, the integrins α2β1, α5β1, and α6β1 can mediate the binding of unactivated platelets to the subendothelial matrix components collagen, fibronectin, and laminin, respectively, at least in vitro.15 However, as discussed in Chapters 28 and 29, at the higher shear rates present in arteries, arterioles, and the microcirculation, efficient platelet adhesion requires the additional presence of subendothelial von Willebrand factor (vWF).16,17 vWF is an elongated multimeric glycoprotein synthesized by endothelial cells and megakaryocytes that binds to the GPIb-IX-V complex on unactivated platelets and to the integrin αIIbβ3 on activated platelets.18 Although vWF is present in plasma, it is normally not present on the platelet surface. However, exposing vWF in vitro to the antibiotic ristocetin or to the snake venom protein botrocetin induces vWF binding to GPIb-IX-V.19,20 As discussed in Chapter 26A, the factors that enable platelets to
adhere to vWF in vivo are likely shear stress-induced changes in the conformations of vWF, GPIb-IX-V, or both and/or changes in the conformation of vWF that occur when it is incorporated into the subendothelium.21,22 Regardless, the bonds between vWF and GPIb-IX-V are characterized by rapid on and off rates so that platelets tend to translocate along, rather than firmly adhere to, vWF-coated surfaces.23,24 This reduces platelet velocity, facilitating subsequent adhesion and activation events that would otherwise not occur under high-flow velocity conditions.
adhere to vWF in vivo are likely shear stress-induced changes in the conformations of vWF, GPIb-IX-V, or both and/or changes in the conformation of vWF that occur when it is incorporated into the subendothelium.21,22 Regardless, the bonds between vWF and GPIb-IX-V are characterized by rapid on and off rates so that platelets tend to translocate along, rather than firmly adhere to, vWF-coated surfaces.23,24 This reduces platelet velocity, facilitating subsequent adhesion and activation events that would otherwise not occur under high-flow velocity conditions.
Collagen is not only a substrate for platelet adhesion, but it is a binding site for vWF in the subendothelium and an agonist for platelet aggregation and secretion, as discussed in Chapters 28 and 30. Although a number of platelet proteins have been found to interact with collagen, only two, the immunoglobulin superfamily member glycoprotein VI (GPVI)25 and the integrin α2β1 (also known as GPIa-IIa),26 are physiologic platelet receptors for collagen. As currently understood, the relatively low affinity interaction of GPVI with collagen, mediated by the more distal of its two extracellular Ig-C2-like domains27 and glycine-proline-hydroxyproline triplets in collagen,28 initiates a signal transduction activation pathway via the Fc receptor γ chain noncovalently associated with GPVI.29 This eventually results in the activation of phospholipase Cγ 2 and the hydrolysis of the relatively scarce membrane phospholipid phosphatidylinositol 4,5-bisphosphate into diacylglycerol (DAG) and inositol 1,4,5-triphosphate (IP3) to activate protein kinase C (PKC) and increase the intracellular concentration of Ca2+, respectively.30 In turn, the latter increase the affinity of α2β1 for glycine-x-hydroxyproline-glycine-glutamic acid-arginine (GxOGER) motifs, enabling activated platelets to adhere firmly to collagen.31,32
Platelet Aggregation
Following adhesion, platelets aggregate to form hemostatic plugs or platelet-rich thrombi. In contrast to platelet adhesion, platelet aggregation is an active metabolic process in which agonist binding to specific membrane receptors initiates signaling pathways that enable the integrin αIIbβ3 (GPIIb-IIIa) to bind soluble macromolecular ligands such as fibrinogen or vWF.33 The fibrinogen and/or vWF bound to αIIbβ3 then crosslinks platelets into a hemostatic plug or thrombus. The events responsible for platelet aggregation are summarized in Chapters 26B and 31.
αIIbβ3, the platelet structure directly responsible for platelet aggregation, is a calcium-dependent, noncovalent heterodimer composed of two type I transmembrane glycoproteins, αIIb and β3.33 Each glycoprotein contains a large extracellular domain, a single transmembrane domain, and a short cytoplasmic domain that can interact with a variety of cytoskeletal and signaling proteins.34 There are approximately 80,000 copies of αIIbβ3 per platelet, making it the most densely expressed platelet membrane protein.35 Based on the estimated platelet surface area, αIIbβ3 molecules are spaced <200 Å apart. Rare congenital deficiency or dysfunction of αIIbβ3 produces the autosomal recessive bleeding disorder Glanzmann thrombasthenia (GT).36 Patients with GT usually present in infancy with mucocutaneous bleeding that can be severe.
Crystal structures for the extracellular domains of αIIbβ3 and the related integrin αvβ3 indicated that these domains are bent over on themselves when the integrins are inactive; accompanying electron microscope images suggested that the molecules are extended when the integrins are active.37,38,39 Ligand binding to αIIbβ3 requires that its extracellular domain shifts from a closed low-affinity to an open high-affinity conformation, the end result of a concerted “switchblade-like” rearrangement of the entire heterodimer that begins with separation of the cytoplasmic and transmembrane domains.38 The resting conformation of αIIbβ3 appears to be maintained by an extended interface involving portions of its cytoplasmic, transmembrane, and extracellular stalk domains.40 αIIbβ3 activation occurs when this interface is disrupted by binding of the cytoskeletal proteins talin and kindlin-3 to the β3 cytoplasmic domain.41 In turn, this causes transmembrane and stalk domain separation and the exposure of ligand-binding sites on the αIIbβ3 extracellular domain (FIGURE 22.2).
The major αIIbβ3 ligand is fibrinogen.33 Fibrinogen is composed of pairs of Aα, Bβ, and γ chains folded into three nodular domains. Although peptides corresponding to the carboxyl terminal 10 to 15 amino acids of the γ chain or containing the arginine-glycine-aspartic acid (RGD) motif present at two places in the α chain can inhibit fibrinogen binding to αIIbβ3, fibrinogen binding is mediated by the γ chain sequences.42,43 Nevertheless, clinical studies confirm that RGD-containing peptides inhibit αIIbβ3 function.44 Based on the three-dimensional structure of fibrinogen, the ability of RGD-based peptides and peptidomimetics to inhibit αIIbβ3 function would seem paradoxical, but crystal structures of RGD and γ chain peptides bound to αIIbβ3 revealed that the RGD and γ chain-binding sites on αIIbβ3 overlap.45
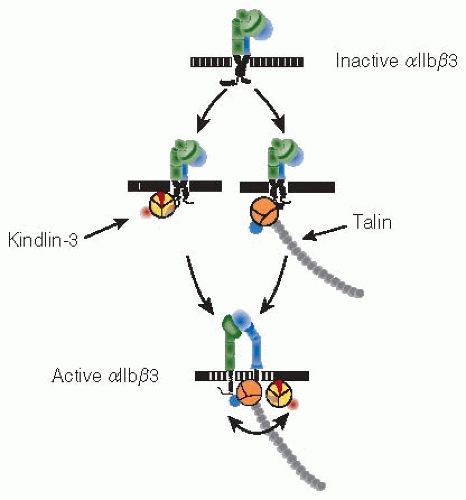 FIGURE 22.2 Activation of αIIbβ3 by talin and kindlin. Cartoon illustrating that both talin and kindlin-3 must bind to the β3 cytosolic domain to cause αIIbβ3 activation by inside-out signaling. Whether there is a preferred sequence of protein binding to β3 remains to be determined. (Adapted from Moser M, Legate KR, Zent R, et al. The tail of integrins, talin, and kindlins. Science 2009;324:895-899, with permission.) |
Platelet Agonists
Agonist-stimulated platelet activation is required to enable binding of soluble ligands to αIIbβ3. As discussed in Chapter 33, the receptors responsible for platelet activation by the classical platelet agonists have all been identified. The serine protease thrombin is the most potent platelet agonist. Human platelets contain two signaling receptors for thrombin, PAR1, and PAR4.46,47 Although PAR1 and PAR4 are members of the large family of GTP-binding protein (G-protein)-coupled seven transmembrane domain receptors, they are unique in that they are activated when thrombin cleaves their amino-termini to expose new termini that act as tethered intramolecular ligands.48,49 Tethered ligand binding, in turn, activates the G-proteins Gq, G12/13, and Gi to stimulate phospholipase Cβ2 activity, increase cytosolic calcium, induce platelet shape change, and inhibit platelet adenylyl cyclase. Since higher thrombin concentrations are required to cleave PAR4 than PAR1, it is likely that PAR1 is the predominant receptor for thrombin at the low thrombin concentrations needed to activate platelets, but once activated, PAR4 signaling is more prolonged than PAR1.50,51
ADP, the first platelet agonist to be identified, is secreted by stimulated platelets from storage sites in platelet-dense granules and may be released from damaged tissue and red cells. There are at least three platelet receptors for ADP.52 The first, P2X1, is an ATP-gated nonselective Ca2+ channel whose physiologic function in platelets is unclear, although P2X1 stimulation by ATP in vitro can amplify the platelet response to low concentrations of other agonists.53 The other two receptors are G-protein-coupled seven transmembrane domain proteins. ADP binding to P2Y1 activates phospholipase Cβ via the G-protein Gq and induces platelet shape change, perhaps via the G-protein G12/13.52,54 ADP binding to P2Y12 inhibits platelet adenylyl cyclase via the G-protein GI.52 P2Y12 is the pharmacologic target for clopidogrel and other thienopyridines.52 Simultaneous stimulation of both P2Y1 and P2Y12 is required to induce platelet aggregation.54,55 Moreover, because antagonism of either the Gq– or Gi-coupled ADP receptor can be overcome by stimulating another platelet receptor coupled to Gq or GI, respectively, it appears that signals emanating each G-protein are needed to activate αIIbβ3.56
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Molecular Basis for Platelet Secretion
Molecular Basis for Platelet Secretion
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


