The focus in Section A of Part III of this book is on inherited hemorrhagic disorders that result from deficiencies or abnormalities of the blood coagulation factors. Acquired hemorrhagic disorders, such as liver disease and diffuse intravascular coagulation (DIC), are presented in Part V of the text. Bleeding disorders from platelet defects are presented in the section on platelets (Section C, Part III) and bleeding disorders due to disorders that affect blood vessels are presented in the section on vascular defects (Section D, Part III). Altogether, worldwide, inherited hemorrhagic disorders due to deficiencies or abnormalities of blood coagulation factors number approximately 250,000. Thus, these are relatively rare but clinically important disorders.
Inherited disorders of blood coagulation have been known since antiquity. FIGURE 49.1 illustrates a passage from the Babylonian Talmud.1 Rabbi Judah the Patriarch, redactor of the Mishnah, the second century compendium of Judaism’s oral law, writes this passage which translates to: “If she circumcised her first child and he died, and a second one also died, she must not circumcise her third child.” This demonstrates knowledge of the inherited nature of the bleeding disorder we now know as hemophilia and also demonstrates knowledge of the sex-linked nature of the disease as well as the clinical penetrance. General knowledge of inherited bleeding disorders such as hemophilia was further amplified when the daughters of Queen Victoria of England, a silent carrier for hemophilia A, married into a number of royal houses in Europe, including Russian, German, and Spanish royalty. The story of Princess Alexandra and her hemophilic son, Alexis, who was cared for by the mad monk Rasputin, is well known and was chronicled by Robert Massie, who was himself the father of a hemophilic son.2
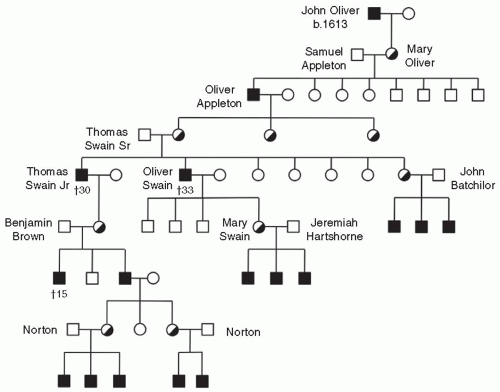
Hemophilia in America goes back a long way too—relatively speaking—to the early colonial days in New England (FIGURE 49.2). John Oliver, the first known hemophiliac in the new world, was born in 1613 in London and came to Plymouth, MA, in 1639 on the ship, Jonathan, eventually settling in Ipswitch, MA. A daughter, Mary Oliver, was born in 1640. In 1656, Mary, an obligate carrier for hemophilia A, was married to Samuel Appleton and they proceeded to have eight children, three daughters and five sons. One son, Oliver Appleton, born in 1677, had hemophilia. Oliver lived to an advanced age and had three daughters. The oldest married Thomas Swain, Sr., of Reading, MA, and had seven children, two boys and five girls. The two sons both had hemophilia and bled frequently but became physicians, lived into their 30s, and had families. Dr. Thomas Swain, Jr., had a daughter who had three sons, two reported to be bleeders. Dr. Oliver Swain had three normal sons and a daughter who had three sons, all with hemophilia. A daughter of Thomas Swain, Sr., married John Bachilor and had three sons with hemophilia. Dr. Oliver Swain’s daughter, Mary Swain, married Jeremiah Hartshorne in 1786 and gave birth to three sons with hemophilia. The Hartshorne family lived in Wakefield, MA, and, as it happened, next door to Dr. John Hay, a fellow of the Massachusetts Medical Society. Upon learning of the family history and after caring for the Hartshorne children, Dr. Hay published an Account of a Remarkable Hemorrhagic Disposition Existing in Many Individuals of the Same Family in the New England Journal of Medicine in 1813.3,4 Coincidentally, a descendent of Thomas Swain, the New England landscape artist R. Swain Gifford, is an ancestor of the brother-in-law of one of the authors of this text.
Inherited disorders of blood coagulation range from the relatively common von Willebrand disease (vWD) to rare disorders such as factor XIII deficiency and multiple coagulation factor deficiencies (MCFD), from autosomal to X-linked in inheritance, and from severe to mild in clinical manifestations. Table 49.1 presents some of the distinguishing characteristics of each. The differential diagnosis of hemorrhagic disorders, both clinical and laboratory aspects, is discussed in Chapter 50. Specific coagulation tests are available for each factor, making unequivocal diagnosis of inherited deficiencies relatively easy, and industries to procure and sell specific clotting factordeficient plasmas for specific assays exist, providing standardized substrate for the assays. Both immunologic and functional assays are available for most factors, enabling the diagnosis of both quantitative, defined as a proportional reduction in clotting factor protein and activity, and qualitative, defined as a reduction in clotting factor activity in excess of protein, disorders. The basis for clotting factor assays and how preanalytical variables can affect their accuracy is described in detail in Chapters 50 and 58. In general, the assays are sensitive to clotting factor activity, especially to levels of 20% of normal and lower, and are specific, making the diagnosis of most cases relatively straightforward. Where the diagnosis becomes difficult is with mild deficiencies, when clotting factor levels begin to blur with normal individuals, and in cases of general inhibitory substances, such as anti-phospholipid or so-called lupus-like antibodies, where multiple clotting factors may appear to be deficient. For example, the current difficulty with the diagnosis of mild type 1 vWD is well known and is discussed in detail in Chapter 52 by Sadler and Lillicrap on vWD.
While deficiency of procoagulants leads to bleeding, the clinical symptoms vary considerably (Table 49.1) and may provide a clue to the diagnosis. Hemophilia A and B are the most severe of the inherited bleeding disorders and have a curious predilection for bleeding into joints that is still poorly understood. As described in Chapter 51, this recurrent bleeding in joints leads to a chronic and progressive form of arthropathy that characterizes the disorder and has some similarity to rheumatoid arthritis. The pathophysiology of the inflammatory changes that lead to the chronic arthropathy is reasonably well understood, but why bleeding occurs in the joints remains a mystery. It has been stated that factors VIII and IX must have some function supporting joint vessels. Bleeding from delicate mucous membranes is a feature of von Willebrand factor (vWF) deficiency (Chapter 52), suggesting that vWF-mediated platelet adhesion is particularly important in these tissues. Joint bleeding may occur in vWD but usually correlates with the level of factor VIII deficiency. Factor XIII deficiency has a high rate of intracranial bleeding, leading clinicians to emphasize the importance of prophylaxis in this disorder. Surprisingly, complete deficiency of fibrinogen (Chapter 54) produces a less serious bleeding disorder than might be expected from the deficiency of the protein that actually produces the clot. Why don’t patients with afibrinogenemia bleed continuously? This is a question that is frequently asked by students and practitioners. In newborns with afibrinogenemia, bleeding from the umbilical stump is common. In children and adults, bleeding is typically in soft tissues and organs, but joint bleeding is not common. Other deficiencies (Chapter 53) are less common, and some, like factor XI deficiency, may be more variable in their bleeding but no less important if encountered. Despite the significant prolongation of the partial thromboplastin time (PTT) that occurs with deficiencies of factors XII, high molecular weight kininogen, and prekallikrein, there is no bleeding tendency. These proteins are important, though, providing, along with activated protein C, connections between the coagulation and inflammation pathways.
FIGURE 49.1 Hemophilia in Antiquity. A familial bleeding disorder, probably hemophilia, is described in the Babylonian Talmud, written by Rabbi Judah the Patriarch, redactor of the Mishnah, in the second century compilation of Jewish law. The translation is: “For it was taught: If she circumcised her first child and he died, and a second one who also died, she must not circumcise her third child. However, Rabbi R. Simeon b. Gamaliel said: She circumcises the third, but must not circumcise the fourth child.”
FIGURE 49.2Hemophilia in the New World. Pedigree of first reported family with hemophilia in North America, as reported by Hay in 1813.3 (The figure is reconstructed from descriptions in Hay’s publication, in part.)
Table 49.1 Inherited hemorrhagic diseases
Diagnostic Testing
Defect
Frequency
Inheritance Pattern
Bleeding Manifestations
PT
PTT
TCT
BT
Fibrinogen abnormalities
Afibrinogenemia
Autosomal
Severe, but less so than severe hemophilia A and B
Infinite
Infinite
Infinite
Prolonged
Dys-fibrinogenemia
Autosomal
Variable bleeding and/or clotting
Prolonged
Prolonged
Prolonged or shorten-ed
Normal
Prothrombin deficiency
Autosomal
Varies with prothrombin levels
Prolonged
Prolonged
Normal
Normal
Factor V deficiency
Autosomal
Mild-moderate
Prolonged
Prolonged
Normal
Prolonged
Factor VII deficiency
Autosomal
Moderate-severe
Prolonged
Normal
Normal
Normal
Hemophilia A
X-linked recessive
Variable, depending on factor VIII level
Normal
Prolonged
Normal
Normal
Hemophilia B
X-linked recessive
Variable, depending on factor IX level
Normal
Prolonged
Normal
Normal
Factor X deficiency
Autosomal
Variable, depending on factor X level
Prolonged
Prolonged
Normal
Normal
Factor XI deficiency
Autosomal
Variable, but not dependent on Factor XI levels
Normal
Prolonged
Normal
Normal
Deficiency of factor XII
Autosomal
None
Normal
Prolonged
Normal
Normal
Deficiency of prekallikrein
Autosomal
None
Normal
Prolonged
Normal
Normal
Deficiency of high molecular weight kininogen
Autosomal
None
Normal
Prolonged
Normal
Normal
Factor XIII deficiency
Autosomal
Severe
Normal
Normal
Normal
Normal
vWD
1:1,000,000
Autosomal; usually recessive; more rarely dominant
Moderate to mild, depending on level of deficiency and defect; predilection for mucous membranes
Normal
Normal or prolonged
Normal
Normal or prolonged
Deficiency of α2 plasmin inhibitor
Autosomal
Moderate
Normal
Normal
Normal
Normal
Deficiency of PAI -1
Autosomal
Moderate
Normal
Normal
Normal
Normal
Only gold members can continue reading. Log In or Register to continue

 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient



