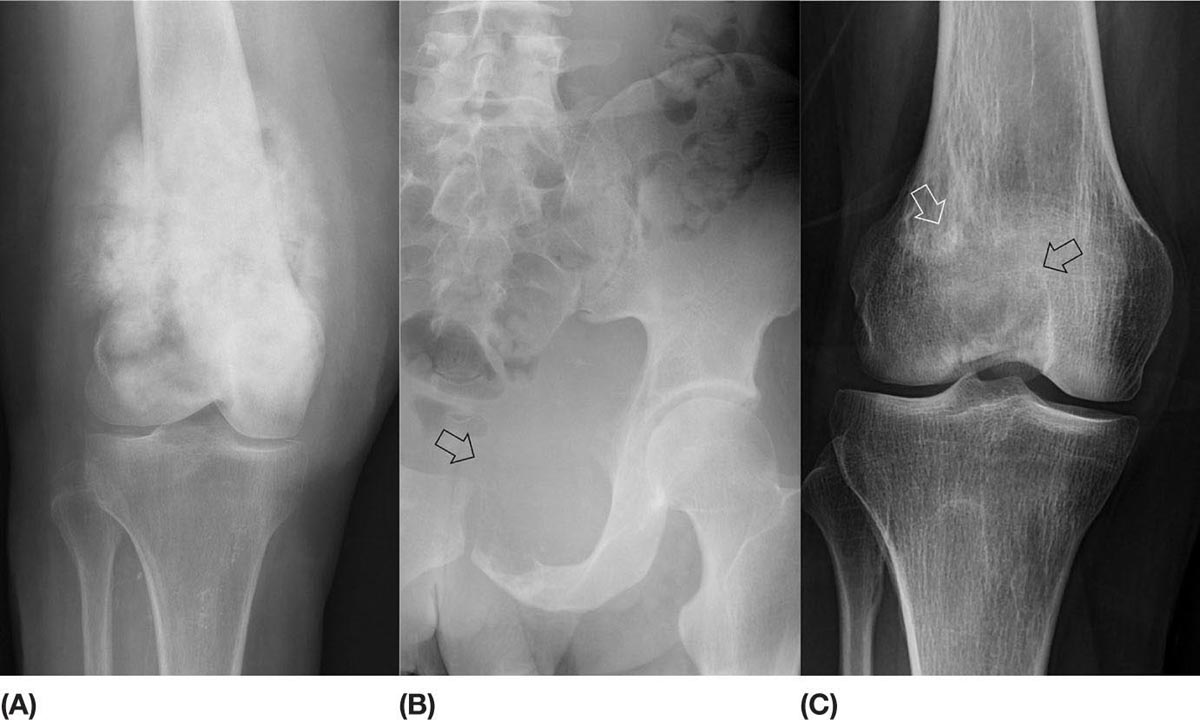
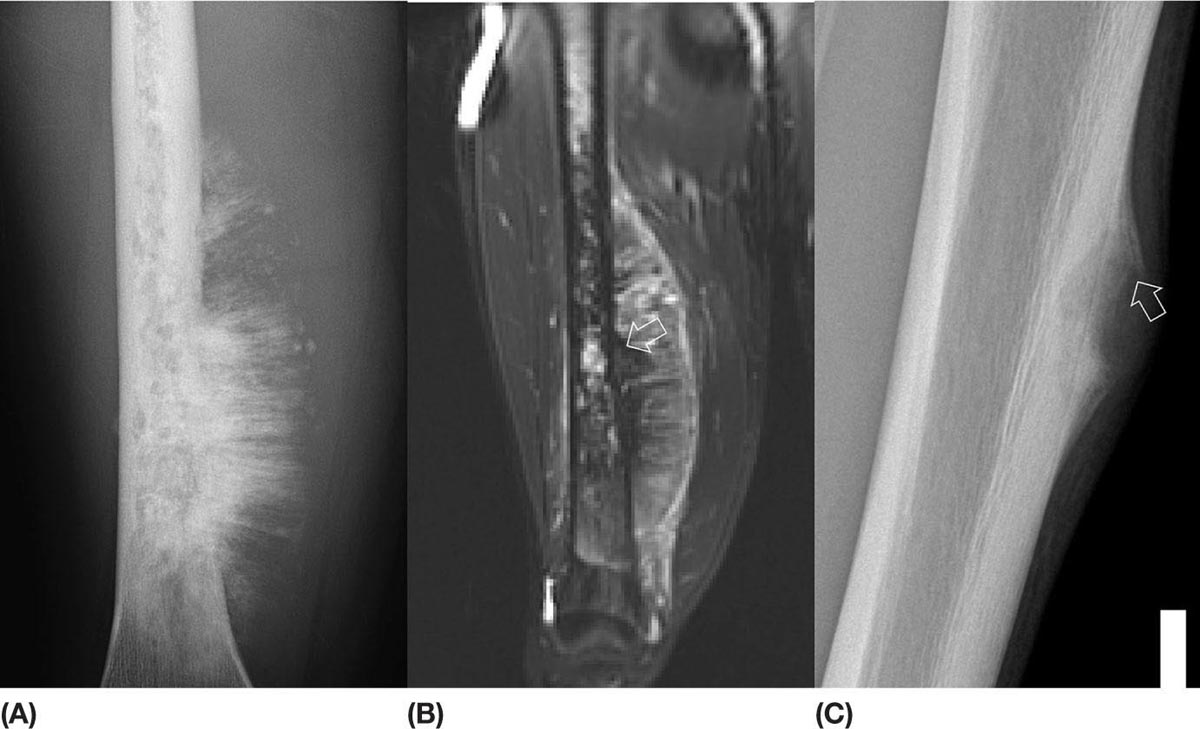
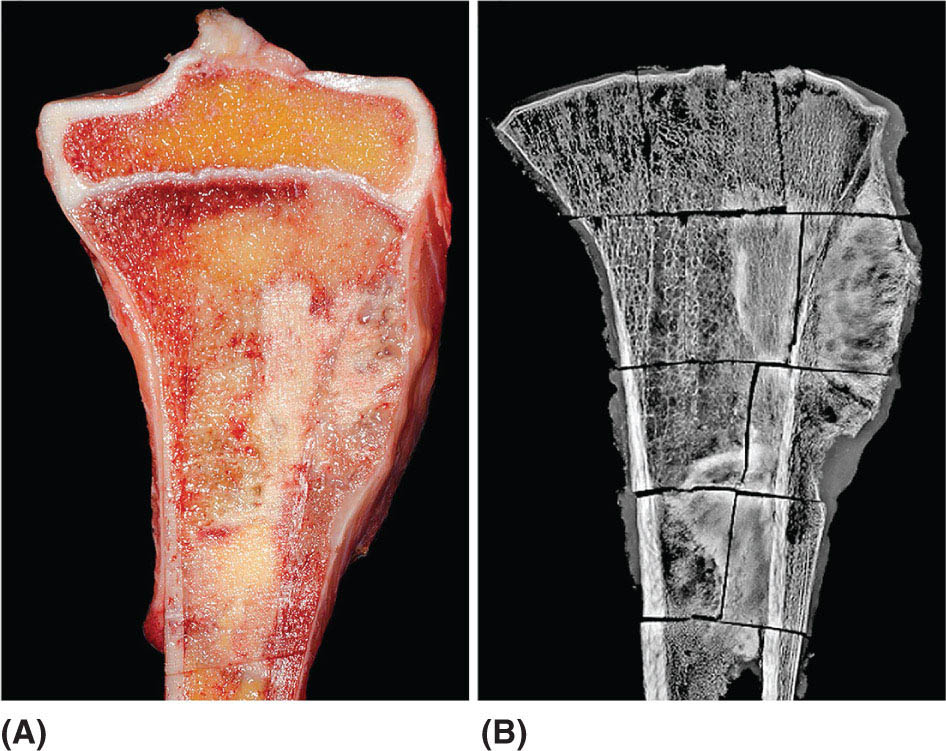
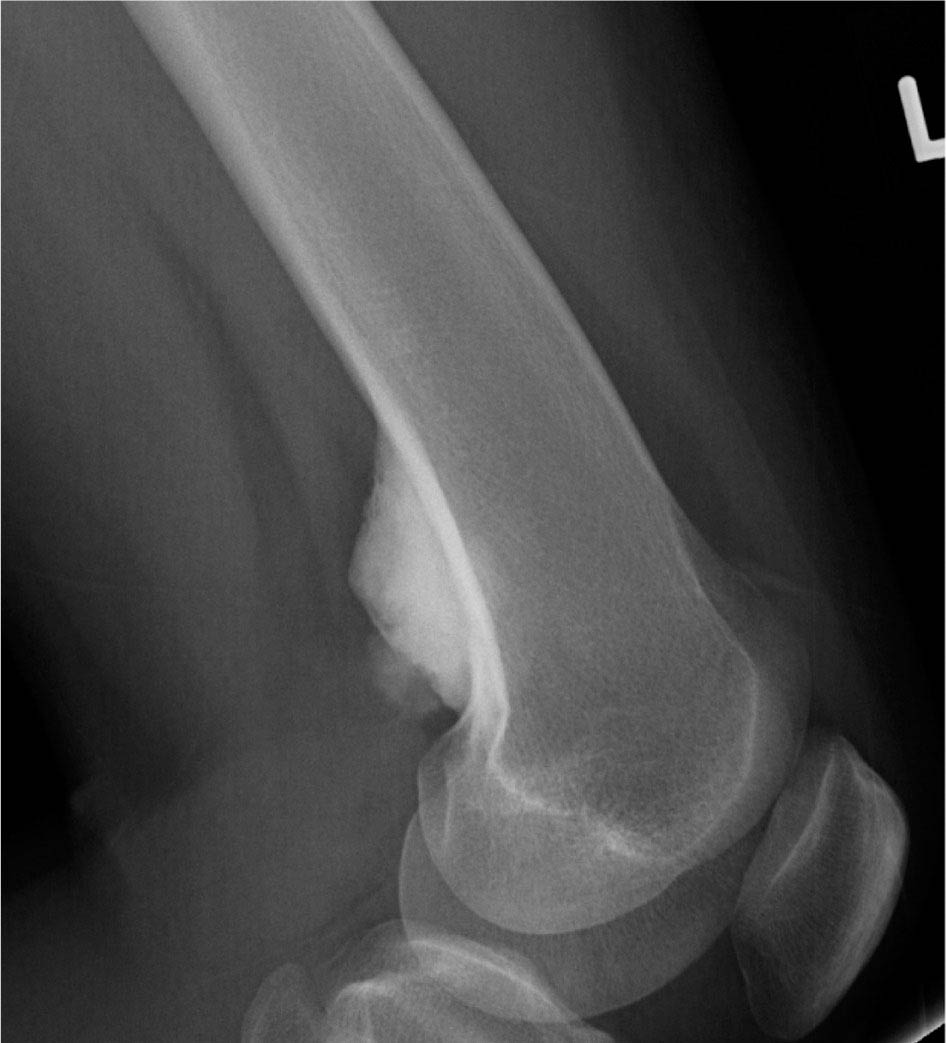
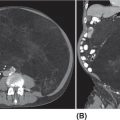
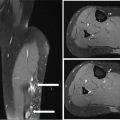
14911 Osteosarcoma Osteosarcoma is the most common primary bone sarcoma. Although osteosarcoma previously held a poor prognosis, overall survival improved dramatically after the adoption of multiagent chemotherapy, establishing multimodality treatment including surgery and chemotherapy as the standard of care. Since these advances over 30 years ago, overall treatment strategies for the primary treatment of osteosarcoma have not changed significantly, and therefore, overall survival has remained relatively stable. This chapter reviews incidence, epidemiology, presentation, and diagnostic approaches for osteosarcoma, as well as general therapeutic approaches for both primary tumor and metastatic disease. The mainstay of treatment involves neoadjuvant chemotherapy, wide surgical resection with limb-sparing surgery if possible, followed by adjuvant chemotherapy. The treatment of metastatic disease remains a significant challenge. While patients with isolated pulmonary metastases may be cured with resection, unresectable disease portends a poor prognosis. Current approaches being developed include defining molecular subsets for targeted therapies and exploring the role of immunotherapy for the treatment of recurrent/metastatic disease. osteosarcoma, neoadjuvant/adjuvant chemotherapy, limb salvage surgery adjuvant chemotherapy, immunotherapy, limb-sparing surgery, metastatic disease, multiagent chemotherapy, osteosarcoma, prognosis, standard of care, surgical resection Chemotherapy, Adjuvant, Drug Therapy, Immunotherapy, Limb Salvage, Neoplasm Metastasis, Osteosarcoma, Prognosis, Standard of Care INTRODUCTION Osteosarcoma is the most common primary bone sarcoma, most often affecting adolescents and young adults or arising as a secondary osteosarcoma in older adults. Although osteosarcoma previously held a poor prognosis, overall survival improved dramatically after the adoption of multiagent chemotherapy, establishing multimodality treatment including surgery and chemotherapy as the standard of care. Since these advances over 30 years ago, overall treatment strategies for the primary treatment of osteosarcoma have not changed significantly, and therefore, overall survival has remained relatively stable. The mainstay of treatment involves neoadjuvant chemotherapy (most frequently, a combination of doxorubicin, cisplatin, high-dose methotrexate with or without ifosfamide), wide surgical resection with limb-sparing surgery if possible, followed by adjuvant chemotherapy. The treatment of metastatic disease remains a significant challenge. While patients with isolated pulmonary metastases may be cured with resection, unresectable disease portends a poor prognosis. Current approaches being developed include defining molecular subsets for targeted therapies and exploring the role of immunotherapy for the treatment of recurrent/metastatic disease. INCIDENCE AND EPIDEMIOLOGY Osteosarcoma follows a bimodal distribution, with a peak incidence in children and adolescents and a secondary peak in the elderly. According to the Surveillance, Epidemiology, and End Results Program (SEER) from the National Cancer Institute, the annual incidence in the United States is 4.4 per million for those 0 to 24 years of age, 1.7 for those 25 to 59 years of age, and 4.2 for adults older than 60 years.1 Among adolescents and young adults, the majority of cases are conventional high-grade osteosarcoma (including osteoblastic, chondroblastic, and fibroblastic subtypes). Conversely, older adults have higher rates of osteosarcoma associated with Paget disease of bone or as a secondary cancer.1 Overall, there is also a slight predominance in blacks and males, with an average male/female ratio of 1.22:1.1 Several studies have examined the prognostic factors and survival outcomes in the current treatment era of multimodality therapy. The recently completed European and American Osteosarcoma Study (EURAMOS-01), an international collaboration of the largest osteosarcoma trial performed to date, reported the 3- and 5-year overall survival rate from the date of diagnosis as 79% and 71%, respectively, in a cohort of patients aged ≤40 years with resectable high-grade osteosarcoma.2 The 3- and 5-year event-free survival rates in this cohort were 59% and 54%, respectively. Factors associated with adverse outcome included both pulmonary and nonpulmonary metastases upon presentation, axial skeleton tumor site, and poor histologic response after chemotherapy (defined as ≥10% viable tumor), with poor histologic response being the most significant prognostic adverse factor.2 Analysis across multiple cooperative group trials and registry-based studies have identified similar prognostic factors. The most important prognostic factor at the time of diagnosis is the presence of detectable metastatic disease. Survival outcomes for the 15% to 20% of patients who present with overt metastatic disease are poor, with a 10-year cause-specific survival rate of 24% as compared to 66% for patients with localized disease.3 Male sex, axial tumor site, large tumor size, and older age at diagnosis have also been associated with inferior outcomes.3,4 Following neoadjuvant chemotherapy, pathologic treatment response—with poor response often defined as <90% tumor necrosis in the primary tumor—correlates with both event-free and overall survival.4,5 The prognosis for patients with secondary osteosarcomas, such as radiation-induced and pagetoid osteosarcoma, is poor, with overall survival rates at 5 years between 1504% and 22%, likely due to a combination of chemoresistance, disease biology, and poor tolerance of standard chemotherapy in the elderly population.6–8 ETIOLOGY The specific molecular pathogenesis of osteosarcoma remains unclear. Although the vast majority of cases occur sporadically, its occurrence has been classically linked to germline alterations in tumor suppressor genes implicated in several inherited familial cancer predisposition syndromes, such as germline mutations in the RB1 gene in retinoblastoma, TP53 gene in Li–Fraumeni syndrome, and RECQL4 gene in Rothmand–Thomson syndrome.9–11 A small, but clinically important subset of cases is caused by exposure to therapeutic radiation with an average latency period of >10 years between irradiation and the development of secondary osteosarcoma. In older adults, osteosarcoma is often associated with Paget disease of bone. However, the incidence of malignant transformation is <1%. Unlike other sarcomas, osteosarcomas lack recurrent translocations or common driver mutations. Rather, osteosarcomas are characterized by complex karyotypes and genomic complexity attributed to mutations in the tumor suppressor genes TP53 and RB. Initially thought to account for approximately half of osteosarcomas, now the vast majority (75%–95%) of all are estimated to harbor TP53 alterations, frequently translocations in intron 1 of the TP53 gene.12 In addition to TP53 mutations, chromosomal shattering, termed chromothripsis, is evident in 20% to 89%, contributing further to its genomic complexity.13 More recent studies utilizing next-generation sequencing (whole-exome sequencing [WES] and whole-genome sequencing [WGS]) reveal a genomic landscape predominantly characterized by somatic copy number alterations (SCNAs) rather than somatic mutations. While the highest among pediatric solid tumors, the somatic mutation burden in osteosarcoma (0.79–1.3 per Mb) is modest in comparison to other adult-type solid tumors.12,14 PRESENTATION AND DIAGNOSTIC APPROACH Patients most often present with deep-seated, constant pain and swelling in the affected location. As the most common locations are in the metaphysis of long bones, specifically the distal femur, proximal tibia, and proximal humerus, they will often endorse knee or shoulder pain or limited range of motion and attribute it to minor trauma, a sprain, or overuse injury. In some instances, patients may even present with pathologic fracture necessitating immobilization or provisional stabilization. Occasionally, patients may present with back or pelvic pain as well, as approximately 6% of cases occur in the axial skeleton.4 Fortunately, these cases are uncommon, as they are typically the most challenging to resect surgically and often are not recognized until late in the disease course. In addition to a detailed history and physical examination, the workup for osteosarcoma includes laboratory tests, radiographs, MRI of the entire bone to evaluate for skip lesions (i.e., discontinuous metastases in the primary bone site), technetium bone scan, and chest CT. Chest imaging is essential as the most common site of metastasis is the lungs. Although not universally accepted as the standard of care, whole body (i.e., head-to-toe) PET/CT is also often performed, which provides additional staging information and may aid in assessing treatment response. Favorable PET/CT responses are associated with improved progression-free survival (PFS), but this only partially correlates with the histologic response to chemotherapy.15 Radiographic evaluation demonstrates an osteoblastic, osteolytic, or mixed lesion (Figure 11.1A–C) and also provides a measure by which treatment response may be evaluated. The classic appearance is a permeative, moth-eaten, or destructive lesion in bone with an associated ossified or “sunburst” appearance of calcification (Figure 11.2A and B). Codman triangle (Figure 11.2C), a periosteal calcification in the border of the tumor and healthy tissue that occurs as a reactive process to wall off the tumor, is also commonly seen on radiographs (Figure 11.3A and B). Secondary osteosarcomas can have a similar appearance with a calcified mass arising from Paget disease or within a prior radiation field (Figure 11.4A and B). MRI remains the best modality with which to evaluate soft tissue component, surrounding neurovascular structures, and intramedullary extension—features that can be valuable in surgical planning. While imaging guidelines for the workup, treatment, and follow-up of osteosarcoma patients differ slightly among the National Comprehensive Cancer Network (NCCN), European Society for Medical Oncology (ESMO), and the Children’s Oncology Group (COG), the same imaging should be utilized during treatment and for posttreatment disease surveillance. FIGURE 11.1 Variable appearance of osteosarcoma. (A) Typical blastic appearance. (B) Lytic appearance of small cell osteosarcoma. (C) Mixed lytic and blastic appearance. Source: Used with permission from Behrang Amini, MD, Department of Diagnostic Radiology, The University of Texas MD Anderson Cancer Center. Laboratory testing should include a complete blood count, electrolyte panel, blood urea nitrogen, creatinine, and liver function tests. Alkaline phosphatase and lactate dehydrogenase levels have also been described as a surrogate measure of tumor burden, with elevated levels correlating with poor prognosis,16 but are not routinely followed as tumor markers. Pretreatment workup should also include echocardiogram and fertility counseling for patients with high-grade osteosarcoma who will go on to receive chemotherapy. FIGURE 11.2 Periosteal reaction in osteosarcoma. (A) Sunburst appearance in conventional osteosarcoma on conventional radiography. (B) T2-weighted image showing calcification perpendicular to the cortex, lifting the periosteum (thin dark line, arrow). (C) Codman triangle (arrow) in a patient with periosteal osteosarcoma. Source: Used with permission from Behrang Amini, MD, Department of Diagnostic Radiology, The University of Texas MD Anderson Cancer Center. FIGURE 11.3 (A) Gross specimen of osteosarcoma involving metaphysis and diaphysis of tibia. Tumor is predominantly intramedullary and extends through cortex with prominent periosteal reaction. (B) The x-ray image of mapped cross section. Source: Used with permission from Wei-Lien Wang, MD, Department of Pathology, The University of Texas MD Anderson Cancer Center. In addition to radiographic evaluation and staging workup, the diagnosis must always be verified histologically with a biopsy. It is widely accepted and recommended that biopsies should be performed at specialized centers with experience treating sarcomas owing to their rarity, in order to ensure proper biopsy techniques and appropriate histologic examination and molecular testing when indicated. Core-needle biopsies are sufficient, but multiple samples should be obtained to improve diagnostic yield. With the increased accuracy of image-guided needle biopsies, the need for open biopsy has diminished, but it remains the gold standard for obtaining sufficient tissue for diagnosis. When necessary, open biopsies should be performed by experienced orthopedic oncologists. Placing the biopsy incision in line with the incision for definitive resection is essential because the biopsy tract must be excised as well. FIGURE 11.4 Secondary osteosarcoma. (A) Osteosarcoma (white arrow and inset) arising from Paget disease of the left ilium (black arrow). (B) Anterior chest wall osteosarcoma associated with radiation therapy for head and neck cancer. Source: Used with permission from Behrang Amini, MD, Department of Diagnostic Radiology, The University of Texas MD Anderson Cancer Center. FIGURE 11.5 (A) Pretreatment biopsy. High-grade tumor composed of atypical epithelioid cells with osteoid matrix (H&E, 200×). (B) Posttreatment resection specimen. Treatment-related changes result in decreased cellularity and replacement by hyaline stroma. Osteoid matrix remains (H&E, 200×). H&E, hematoxylin and eosin. Source: Used with permission from Wei-Lien Wang, MD, Department of Pathology, The University of Texas MD Anderson Cancer Center. The Musculoskeletal Tumor Society (MSTS) and the American Joint Committee on Cancer (AJCC) systems have been traditionally used for staging.17,18 Most patients present with nonmetastatic conventional high-grade osteosarcoma with soft tissue component, or MSTS stage IIB disease. In the most recent eighth edition of the AJCC classification, which uses the traditional TNM (primary tumor, regional lymph nodes, and distant metastasis) staging of solid tumors, T stage is separated by the primary site, with spine and pelvis primary tumors classified separately from those arising in the appendicular skeleton, trunk, skull, or facial bones. Prognostic stage groups are defined for tumors arising in the appendicular skeleton, trunk, skull, or facial bones based upon T, N, M, and histologic grade. Clinically, neither staging system has been routinely incorporated into treatment planning; rather the presence or absence of overt metastatic disease remains the most significant prognostic factor. Histologic subtypes of osteosarcoma are described in the World Health Organization classification of bone-forming tumors.19 All osteosarcoma subtypes are characterized by osteoid production in a malignant spindle cell stroma (Figure 11.5A and B). Conventional intramedullary osteosarcomas account for the largest subtype of osteosarcoma and are subdivided into osteoblastic, chondroblastic, and fibroblastic subtypes, named according to the predominant cellular component. Other intramedullary histologic subtypes are less common and include telangiectatic and small cell osteosarcoma. Variants such as surface osteosarcomas, which include parosteal, periosteal, and high-grade surface as well as osteosarcoma of the mandible, often differ in regard to natural history, prognosis, and treatment and are discussed in further detail in separate sections of this chapter. Extraskeletal osteosarcoma, a malignant mesenchymal tumor that arises in soft tissue (without bone involvement) and produces osteoid, is considered an entirely separate entity and is most often treated in line with other high-grade soft tissue sarcomas rather than conventional osteosarcoma. GENERAL THERAPEUTIC APPROACH FOR PRIMARY TUMOR The therapeutic approach for osteosarcoma is described in relation to treating conventional, high-grade osteosarcoma. Similar to biopsy principles, treatment is best completed in multidisciplinary referral centers with radiologists, pathologists, orthopedists, and medical oncologists experienced in treating bone sarcomas. Successful treatment requires both systemic chemotherapy and surgical resection. Osteosarcoma is not sensitive to radiation therapy at doses that would allow safe administration of systemic therapy, and therefore is reserved for select cases typically in a palliative setting for patients with incurable disease. Historically, metastatic disease recurrence was high and the long-term survival rate was low (<20%) in patients treated with surgery alone. Comparatively, 5-year overall survival rate 154is >60% for patients treated in the current era with chemotherapy and surgery and >70% for patients with localized osteosarcoma of the extremity.20 These findings indicate the presence of micrometastatic disease in the majority who present with localized disease at the time of diagnosis and support the routine use of chemotherapy in addition to surgery. The most active chemotherapeutic agents include high-dose methotrexate, doxorubicin, cisplatin, and ifosfamide.21,22 While the specific combination or number of chemotherapeutic agents differs across protocols, most chemotherapy regimens use at least three of these four agents. Neoadjuvant chemotherapy is commonly administered as a three-drug combination of high-dose methotrexate, doxorubicin, and cisplatin (MAP). This regimen was agreed upon as the standard therapy for the pediatric EURAMOS-1 trial, based on a prospective, randomized trial performed by the COG.2,23 Repeat imaging at the conclusion of neoadjuvant chemotherapy prior to resection is useful for surgical planning, but has limited utility in assessing treatment response. Histologic response to neoadjuvant chemotherapy measured as percent tumor necrosis (Figure 11.5B) has historically been considered the most significant prognostic factor for disease-free and overall survival. This has been confirmed in contemporary series as well.2,4,24 Patients with <90% tumor necrosis after preoperative chemotherapy have more than double the risk of a subsequent event, whereas those with ≥90% tumor necrosis have a 5-year continuous disease-free survival rate of approximately 80%.2,25 For conventional osteosarcoma, limb-salvage surgery is feasible in >85% of patients without compromising overall survival when compared to amputation.26 If preoperative imaging does not demonstrate extensive neurovascular involvement or a massive soft tissue component precluding safe resection, the limb may be spared and reconstructed using a variety of techniques, including endoprosthesis, allograft–prosthetic composite, or rotationplasty. Intra-articular resection can usually be performed if an MRI does not demonstrate invasion into the joint space or along cruciate or collateral ligaments. In spine and pelvic cases, en bloc spondylectomy or hemipelvectomy are the surgical options depending on the extent of invasion of the epidural space and dura, hip joint, and nearby neurovascular structures. Other surgical considerations include the presence of pathologic fracture, potential remaining skeletal growth in children and resultant limb length discrepancies, and functional demands after the procedure. In order to ensure negative bone margins, frozen sections are taken from the marrow margins and grossly evaluated by the pathologist. Whenever possible, bone margins of 1 to 3 cm are recommended, although subcentimeter margins may be acceptable in epiphyseal-sparing resections when the tumor border is clearly delineated on MRI. The adequate soft tissue margin is more difficult to discern; some studies report local recurrences are significantly decreased using ≥2 mm as a cutoff.27,28 Perhaps more important than an exact cutoff for an adequate margin is the response to chemotherapy, which may modulate aspects of the surgery performed and impact local control. After resection of the primary tumor, patients typically resume adjuvant chemotherapy. While poor histologic response to neoadjuvant chemotherapy is a well-established prognostic factor, the role for modified postoperative chemotherapy for poor responders remains controversial. The concept of “tailoring” chemotherapy has been attempted since at least the 1970s, but early studies were difficult to interpret and relied upon neoadjuvant chemotherapy regimens that are no longer used.29 In subsequent series, the addition of ifosfamide and etoposide (I/E) to postoperative MAP chemotherapy for poor responders resulted in an improvement in disease-free survival.30 These and other studies showing the activity of I/E in osteosarcoma served as the basis for the trial design in EURAMOS-1 study in which patients with poor response to neoadjuvant MAP were randomized to either continue MAP alone or receive intensified postoperative chemotherapy with MAPIE. In this study, MAPIE failed to improve event-free survival among poor responders as compared to MAP and was associated with increased toxicity.31 The treatment of older adults with osteosarcoma warrants special consideration. In most series, older adults have a worse prognosis than children or young adults with osteosarcoma. However, curative intent treatment with combination chemotherapy and resection is warranted when feasible.32 Older adults often have difficulty tolerating the dose and intensity of the pediatric MAP chemotherapy regimen. High-dose methotrexate, in particular, can be difficult to administer in older adults due to the slow clearance of methotrexate and overlapping toxicities of mucositis with doxorubicin and nephrotoxicity with cisplatin.33 At several large sarcoma centers in the United States, adults are treated with neoadjuvant doxorubicin and cisplatin (AP) for up to four cycles, followed by resection and assessment of histologic treatment response. Patients with a favorable pathologic response go on to receive four cycles of adjuvant doxorubicin and ifosfamide (AI), omitting high-dose methotrexate. For patients with poor response, adjuvant chemotherapy consists of six cycles each of high-dose methotrexate and high-dose ifosfamide for up to 12 total cycles. This modified approach has been associated with improved disease-free survival among adults with poor response to neoadjuvant chemotherapy.34 155FOLLOW-UP Strict adherence to long-term follow-up is critical for successful treatment. The NCCN and the COG provide guidelines for general timing for follow-up and disease surveillance depending on the tumor grade and pathologic diagnosis.35,36 Patients with high-grade osteosarcoma should be seen with posttreatment surveillance imaging every 3 months for the first 2 years after treatment, every 4 months in the third year, every 4 to 6 months in the fourth year, and every 6 months in the fifth year. Because late recurrence can be seen >10 years after the completion of treatment, consideration should be given to continuing disease surveillance every 12 months after 5 years. Surveillance imaging consists of radiographs and MRI of the primary bone, as well as chest imaging. Small subcentimeter nodules that are nonspecific are typically followed during surveillance with serial chest CT. Local recurrences are becoming less common with improved surgical technique and imaging, but have been reported in up to 10% of patients. They require wide local excision with negative margins, which sometimes can only be achieved with amputation in cases with extensive involvement.37 After 10 years, the risk of recurrence or secondary malignancy is significantly decreased, but long-term follow-up is still recommended to monitor the status of orthopedic implants and late effects of chemotherapy. Endoprostheses are subject to high rates of wear over time and may eventually fail owing to aseptic loosening, polyethylene wear, hardware breakage, or instability necessitating revision. Allograft constructs may also exhibit nonunion, resorption, or degenerative joint disease requiring total joint arthroplasty. Amputees typically need lifelong follow-up with prosthetists to maintain and adjust their prostheses. Long-term follow-up is also needed to evaluate and treat the frequent treatment-related late effects of chemotherapy. Among childhood cancer survivors, survivors of bone tumors face the highest of risk of developing severe or potentially life-threatening chronic health conditions including renal impairment, cardiomyopathy, hearing loss, and ongoing musculoskeletal problems.38 Osteosarcoma survivors are at increased risk of doxorubicin-induced cardiac dysfunction and cardiomyopathy, which may require regular echocardiograms and evaluation of other cardiac risk factors. Renal impairment due to nephrotoxic chemotherapy such as ifosfamide and cisplatin may present many years after completion of therapy if potentiated by other nephrotoxic medications or other common causes of renal dysfunction such as hypertension, and therefore warrants ongoing monitoring of renal function, electrolytes, and blood pressure. Acute and long-term side effects from cisplatin include sensory neuropathy and ototoxicity; patients should undergo audiology evaluations prior to and during treatment and as clinically indicated. Secondary cancers including treatment-related myelodysplastic syndrome and acute myelogenous leukemia are infrequent but well described and have been specifically linked to alkylating agents and topoisomerase inhibitors.39 THERAPEUTIC APPROACH FOR METASTATIC DISEASE The prognosis for patients with nonmetastatic conventional osteosarcoma has remained relatively constant since the introduction of chemotherapy, with a 5-year overall survival rate of roughly 65% to 75%.2,4 Conventional high-grade osteosarcoma has a high propensity to metastasize, which is clearly evident prior to the introduction of chemotherapy, whereas low-grade variants such as well-differentiated intramedullary and parosteal osteosarcoma generally have low metastatic potential and are typically treated with surgery alone. Patients who present with metastatic disease have a significantly worse prognosis, with reported 5-year survival rates of 30% to 50%.2,40 Approximately 10% to 20% of patients present with metastatic disease, which is most commonly in the lung, but can also manifest in the same bone as a skip lesion or other bones, and very rarely in lymph nodes. The initial preoperative treatment approach is the same as for patients with nonmetastatic disease, and the approach is with curative intent in those with resectable pulmonary metastases. Relapse is seen in approximately 30% of patients with localized osteosarcoma and up to 80% of patients with primary metastatic disease. Importantly, a subset of patients with recurrent metastatic disease following the completion of primary therapy can achieve long-term remission or cure with surgery alone or in combination with additional chemotherapy. Prognosis in the recurrent metastatic setting depends upon the time to recurrence, location of disease, and the ability of achieving a second surgical remission. Among patients previously treated with neoadjuvant chemotherapy and surgery, the 5-year post-relapse survival rate is 35% for patients who relapsed >2 years from the initiation of treatment as compared to 14% for patients whose disease recurred in <2 years.41 Pulmonary metastasectomy results in 5-year overall survival rate of 25% to 40%, even following second, third, or subsequent 156recurrences for patients who achieve surgical remission with no evidence of disease.42,43 For patients with late disease recurrence, surgical resection alone is appropriate. For those patients with short-interval recurrence (i.e., <1 year from primary surgery), extensive, or borderline resectable disease, chemotherapy prior to or following surgery should be considered. While there are no well-controlled data demonstrating a survival benefit to adjuvant chemotherapy following pulmonary metastasectomy, this approach may prolong time to recurrence and potentially may cure. Neoadjuvant chemotherapy for oligometastatic disease has the added benefit of allowing for response assessment by imaging and tumor necrosis at the time of resection. Chemotherapy is the treatment of choice for patients with recurrent unresectable disease where the prognosis remains exceedingly poor. A recent pooled analysis of seven negative Phase 2 trials from the COG and its predecessors demonstrated a 4-month event-free survival rate of only 12% in patients with unresectable disease.44 In addition to retreatment with the standard agents (particularly for those with a long disease-free interval), second-line regimens for recurrent metastatic disease include high-dose ifosfamide with or without etoposide, the combination of gemcitabine and docetaxel, or more recently, tyrosine kinase inhibitors (TKIs).21,45,46 The specific choice of second-line therapy depends upon the patient’s prior treatment and comorbid conditions. While there are no predominant recurrent targetable alterations in osteosarcoma, multitargeted TKIs that target vascular endothelial growth factor receptor (VEGFR) have shown efficacy in the recurrent metastatic setting. Single-agent sorafenib, targeting VEGFR-1, -2, -3, Raf kinase, and platelet-derived growth factor receptor (PDGFR), has modest activity in osteosarcoma with a median PFS of 4-months and a 6-month PFS rate of 29%.47 The addition of the mTOR (mammalian target of rapamycin) inhibitor everolimus to sorafenib also showed clinical activity (median PFS 5 months, 6-month PFS rate 45%), but failed to meet the study’s primary efficacy endpoint targeting a 6-month PFS rate of 50%.48 Regorafenib, an antiangiogenic TKI targeting VEGFRs as well as KIT, RET, RAF, BRAF, PDGFR, and fibroblast growth factor receptor (FGFR), has also shown activity in two recent placebo-controlled Phase 2 trials in metastatic osteosarcoma. In the French REGOBONE study, 17 of 26 (65%) patients treated with regorafenib were progression free at 8 weeks, as compared to zero of 12 patients receiving placebo.49 Findings from the U.S.-led SARC024 study were comparable, with an improvement in median PFS with regorafenib (3.6 vs. 1.7 months).50 Preliminary data for cabozantinib, which targets cMET, VEGFR2, AXL, and RET, also shows activity in patients with metastatic osteosarcoma.51 There has been a long-standing interest in the role of immune response and immunotherapy in osteosarcoma. Liposomal muramyl tripeptide-phosphatidylethanolamine (MTP-PE; also known as mifamurtide), a synthetic bacterial cell wall analog derived from bacille Calmette-Guerin (BCG) that activates macrophages and increases cytokines, was studied in a randomized trial. The addition of mifamurtide to standard MAP chemotherapy improved overall survival, but not event-free survival.23 A subset analysis of patients with primary metastatic disease enrolled on the same study showed a trend toward improvement in overall survival and event-free survival with the addition of mifamurtide; however, these differences were not statistically significant.52 These results led to its approval in Europe, Mexico, and several other countries, but not within the United States, and its role in the treatment of metastatic osteosarcoma remains controverted. As with other solid tumors, there has been a growing interest in the use of immune checkpoint inhibitors in osteosarcoma. In the SARC028 Phase 2 study of pembrolizumab, single-agent anti-PD-1 therapy demonstrated activity in specific soft tissue sarcoma cohorts, with 72% of advanced disease patients with undifferentiated pleomorphic sarcoma or liposarcoma achieving disease control (stable disease or partial response at 8 weeks). However, the response rate in osteosarcoma was low at 5% with disease control at 8 weeks seen in 32% (seven of 22 patients).53 A second study evaluating nivolumab with or without ipilimumab demonstrated higher response rates with the combination among patients with soft tissue sarcomas, but no responses were seen in the two patients with osteosarcoma (one nivolumab alone, other one nivolumab + ipilimumab).54 Based upon these initial clinical trials, further development of immune checkpoint inhibitors in bone sarcoma is focused on novel combinations. LOW- AND INTERMEDIATE-GRADE OSTEOSARCOMA Low-grade osteosarcoma may be intramedullary or parosteal, and is most commonly seen in the posterior aspect of the distal femur (Figure 11.6). Both forms are rare and may be difficult to distinguish from other ossified lesions such as heterotopic ossification, myositis ossificans, osteomyelitis, and other benign bone-forming lesions. Workup of other variants is the same as for conventional osteosarcoma, but treatment for low-grade lesions is wide local resection alone. Prognosis is excellent with reports of over 95% 157survival rate at 5 years, and local recurrence or metastasis is extremely rare, except in the setting of marginal or intralesional resection or areas of dedifferentiation.55,56 Local recurrence is managed with repeat wide excision and close surveillance. Areas of even small foci of dedifferentiation warrant treatment similar to conventional high-grade osteosarcoma with preoperative and postoperative chemotherapy, as dedifferentiated parosteal osteosarcoma is associated with a high risk of metastatic disease recurrence. Limited data exist for the treatment of intermediate-grade osteosarcoma, but the use of conventional chemotherapy comparable to the treatment of high-grade osteosarcoma is generally favored. FIGURE 11.6 Parosteal osteosarcoma. Lateral radiograph showing well-defined osteoid lesion. Source: Used with permission from Behrang Amini, MD, Department of Diagnostic Radiology, The University of Texas MD Anderson Cancer Center.


Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


