Presentation
The most common initial symptom of osteosarcoma is pain.
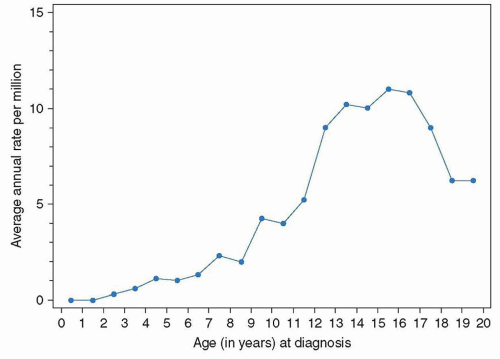
1 This pain can be intermittent but, in general, it becomes continuous and more severe with time. Most patients will relate the onset of symptoms temporally to trauma, but injury is not an etiologic factor in osteosarcoma development per se nor is it clearly tied to any particular event in the tumor’s progression, with the rare exception of a patient who presents with a pathologic fracture. In some patients, a mass may be palpable in the region of the pain, but this is variable. With time, the swelling, if present, will increase and eventually will affect adjacent joints, resulting in loss of function. Given the nonspecific symptoms at the time an osteosarcoma presents, a primary-care provider requires a high index of suspicion to detect the disease early and avoid potentially harmful diagnostic delays and misguided treatment attempts. In assessing the patient, a primary-care provider should consider factors like the age of the patient and the location of the symptoms as these have consistent patterns in osteosarcoma, as will be discussed further in the section on Epidemiology. Unfortunately, these patterns overlap with much more common conditions that afflict this age group. In the typical physically active adolescent, pain caused by osteosarcoma is often initially attributed to recent trauma, commonly related to a sports injury. The differential diagnosis of osteosarcoma also includes osteomyelitis, benign bone tumors such as osteochondroma, fibroma, osteoid-osteoma, enchondroma, giant-cell tumor of bone, bone cysts, and others, as well as other primary malignancies of bone and bone metastases.
2,3Other aspects of how the patient may present can be potentially confusing. For example, pain at an osseous site other than the primary tumor may represent metastatic involvement. That said, the most common site of metastases is the lungs. Respiratory symptoms occur very late and only with the presence of extensive lung involvement. Synchronous (at the time of presentation) and metachronous (following administration of treatment) metastases most commonly involve the lungs. In the two largest reported series of primary metastatic osteosarcomas and metastatic recurrences, pulmonary involvement was observed in 81% and 88%, respectively; the lungs were the only metastatic site in 61% and 70%, respectively.
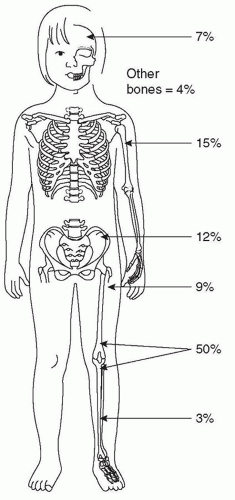
4,5,6 Distant bones are the second most frequent site of metastatic deposits, combined with lung metastases in at least half of all metastatic cases.
5 Skip metastases, isolated tumor foci within the same bone as the primary tumor, represent another form of osseous dissemination.
7 While they occur only in a minority of patients, they must always be sought for by appropriate imaging of the total tumor-bearing bone. Metastases to organs other than lungs or bones are rare in the absence of widespread disease.
5 Weight loss is rare in patients with osteosarcoma and systemic symptoms, such as fever, diaphoresis, and malaise are uncommon in the absence of far advanced disease. Therefore, the vast majority of osteosarcoma patients do not feel systemically ill when their cancer is first diagnosed.
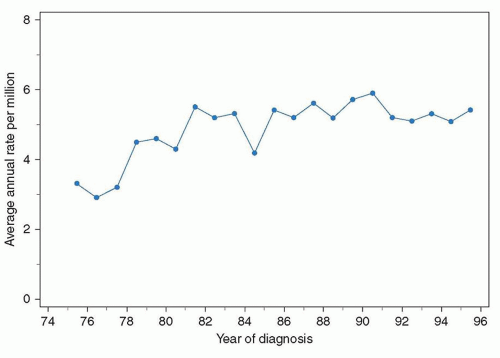
The nonspecific symptoms suggest that in many patients the diagnosis of osteosarcoma may be delayed. The lag-time between onset of symptoms and diagnosis ranges from 2 to 4 months for patients with localized extremity osteosarcoma in several North American and European studies, but it is considerably longer in other parts of the world.
8,9 Despite this frequent delay, there is no correlation between the delay in diagnosis and the likelihood of primary metastases. This would suggest that patients who develop metastases have a biologically more aggressive tumor. Patients with axial primaries are usually diagnosed later than those with tumors located in the limbs.
1,8,9Although osteosarcoma is not often considered the most likely diagnosis in patients presenting with leg pain, arriving at the diagnosis is greatly assisted by the fact that a lesion will invariably be visualized on a plain radiograph. Although imaging is not indicated in every patient with extremity pain, a plain radiograph should be obtained if there is a suspicion for osteosarcoma.
Laboratory Evaluation
There are no known laboratory tests specific for diagnosing osteosarcoma. Serum levels of alkaline phosphatase and less frequently lactic dehydrogenase
10,11 are elevated in a considerable number of patients. Elevated alkaline phosphatase levels do not necessarily correlate with disease extent but have been observed to correlate with an increased likelihood for recurrence.
10,11,12,13 Several other laboratory tests must be performed before beginning chemotherapy. These aim to assess general health, and baseline function for organs affected by chemotherapy treatment. Tests to assess general health include: a complete blood count and differential, tests for serum electrolytes including calcium, magnesium, and phosphate, liver function studies, blood group typing, a coagulation profile, as well as tests for hepatitis and human immunodeficiency virus infection.
Treatment-related complications associated with osteosarcoma treatment include anthracycline-induced cardiomyopathy,
14 hearing loss,
15 kidney dysfunction,
16 secondary malignancies,
17 and sterility especially in patients receiving alkylating agents. These complications serve as an additional guide for baseline tests that should be performed on these patients. In general, these include: an audiogram, an echocardiogram, and renal function evaluation including creatinine and (estimated) creatinine clearance. Additionally, based on the risk of sterility related to chemotherapy, fertility preservation should be considered. Pubertal males are usually referred for sperm donation. Preserving fertility in prepubertal males is currently not feasible, but as technology continues to evolve, referral to a specialist should be considered especially since some institutions may offer experimental testicular tissue cryopreservation. Preserving female fertility is also important and though more complicated, it is necessary to refer female patients to reproductive endocrinology and infertility specialists to review and consider their options.
Radiologic Evaluation
Diagnostic imaging plays a major role in the management of patients with osteosarcoma. In most cases, conventional radiographs suggest the presence of a malignant bone tumor. Further imaging, usually with magnetic resonance imaging (MRI) or computed tomography (CT) of the involved area, is performed to evaluate the extent of local disease. Bone scintigraphy and chest CT scan are also performed to determine the presence and extent of metastatic disease. Additional imaging by 18F-fluoro-2-deoxy-D-glucose (FDG) positron emission tomography (PET), often combined with CT (PET/CT), thallium scintigraphy, or dynamic contrast enhanced MRI (DCE-MRI) may also be performed to further assess the local tumor and, in some cases, to evaluate the response to treatment.
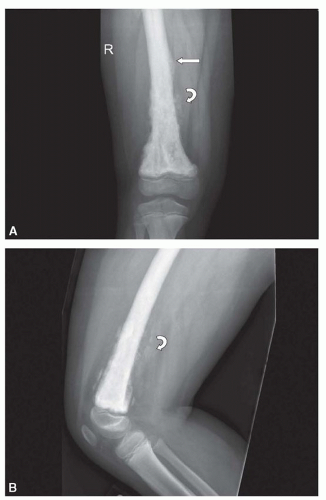
The classic high-grade intramedullary osteosarcoma presents as a large, mixed sclerotic and lucent metaphyseal lesion with fluffy, cloud-like calcific opacities characteristic of osteoid matrix production on conventional radiography (
Fig. 34.1). There is usually a soft tissue mass with aggressive-appearing periosteal reaction, often with a Codman triangle, which results from the soft tissue mass breaking through the overlying ossified periosteal new bone. The periosteal new bone may be spiculated or laminated. Tumors localized in the diaphyses may have onion-skin periosteal reaction similar to Ewing sarcoma. Occasionally, a classic high-grade intramedullary osteosarcoma is completely sclerotic or lucent. The telangiectatic subtype, typically presents as a cystic, lucent lesion with subtle matrix mineralization and a geographic margin with a wide zone of transition.
18 These tumors may be confused with benign aneurysmal bone cysts. CT can be helpful in determining the correct diagnosis by showing subtle matrix mineralization and either CT or MRI may show thick, solid nodular, enhancing tissue surrounding the cystic spaces and the soft tissue mass that are present in a telangiectatic osteosarcoma.
18 Juxtacortical osteosarcomas involve the bone surface and include intracortical, parosteal, periosteal, and high-grade surface lesions. Parosteal osteosarcomas are the most common of these and typically involve the metaphysis of the long bones. These tumors appear radiographically as lobulated masses that attach to the cortex and are ossified centrally. Occasionally, parosteal osteosarcomas must be differentiated from myositis ossificans, which is ossified in the periphery and usually not attached to the cortex. CT is particularly helpful for making this distinction.
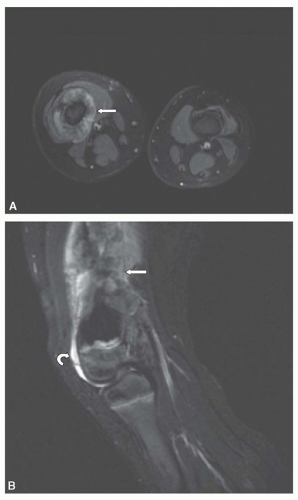
Once a malignant bone tumor is suspected, advanced imaging by CT or MRI should be performed to determine the extent of the tumor within the bone and assess the relationship of the tumor to the nearby neurovascular bundle, muscles, and joints. MRI provides superb contrast resolution and multiplanar capabilities, resulting in images that define the intraosseous and soft tissue components of the tumor and their relationship to adjacent structures (
Fig. 34.2). CT is also able to define the intraosseous and soft tissue components of the tumor and is superior to MRI for the detection of small areas of mineralized matrix. A multi-institutional study
19 found that CT and MRI were equally accurate for local staging without a statistically significant difference between the two imaging modalities for determining local tumor extent. However, MRI is considered the imaging study of choice based on its contrast resolution and the absence of ionizing radiation.
20 Whether CT or MRI is performed for the assessment of the primary tumor, it is recommended that imaging be carried out prior to biopsy. The imaging findings can help in the selection of the optimal site for biopsy and this course of action will avoid distortion of the imaging findings by postbiopsy changes.
21 Imaging of the primary tumor should include coverage of the entire bone to look for the presence of skip lesions. On CT, the extent of intramedullary involvement is determined by the presence of mineralization or soft tissue attenuation material replacing the low attenuation of normal fatty marrow. Intravenous contrast is critical to opacify and help establish the location of the blood vessels. On MRI, tumors are low signal on T1-weighted images and heterogeneous high signal on T2-weighted and STIR (short tau inversion recovery) images. T1-weighted images are more accurate than STIR images in assessing the extent of intraosseous tumor. STIR sequences are very sensitive to fluid and as a result, high-signal peritumoral edema or other benign tissue may simulate tumor (
Fig. 34.2).
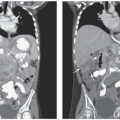
Additional imaging is performed to detect metastatic disease, the majority of which is in the lungs. CT is superior to conventional radiography and is the imaging study of choice to detect
lung metastases.
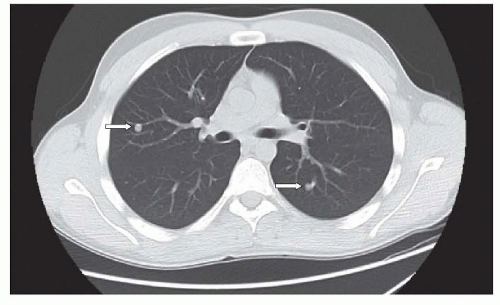
20 In cooperative patients, the CT scan should be performed using spiral technique during a single breathhold with 5 mm or less slice collimation. Unless there is chest wall, hilar, or mediastinal involvement, the CT should be performed without intravenous contrast. In addition, when possible, the CT should be done prior to biopsy to avoid postsedation atelectasis or other postoperative processes that could simulate or obscure lung metastases. Lung metastases are typically round or ovoid, sharply marginated, and located in the lung periphery (
Fig. 34.3). The metastases may or may not be calcified and differentiating benign from metastatic nodules can be difficult, particularly in areas with endemic fungal disease, such as histoplasmosis.
22 The size, number, and margins of the nodules can assist in making this distinction. Sharply marginated lesions larger than 5 mm in diameter, especially when multiple are considered to be possible metastases, while solitary nodules less than 5 mm with unsharp margins are thought more likely to be benign.
23 Though there is no minimum number of nodules that excludes metastases, one study found that all patients with seven or more nodules at surgery had metastases.
24 Following the size of pulmonary nodules, postchemotherapy can be helpful because benign lesions are likely to remain unchanged in size, while lesions that increase or decrease in size are more likely to be metastases.
24 Criteria utilizing nodule size and number have been established to guide the decision (
Table 34.1). In those uncertain cases where the diagnosis is crucial, however, nodule resection and biopsy is the only way to make the distinction. It is also important to note that there are times when granulomas and metastatic disease coexist in the same lesion and patient.
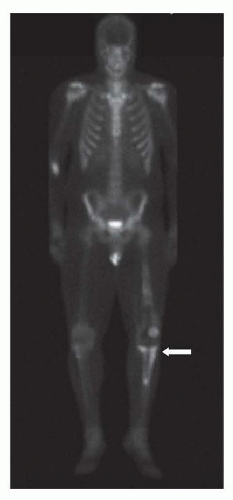
25Bone scintigraphy, typically with technetium-99m-methylene-diphosphonate (MDP) is performed for the assessment of bone metastases and skip lesions (
Fig. 34.4). Conventional radiographs are helpful to assess foci of increased uptake that are suspicious for bone metastases and occasionally MRI is needed to further evaluate these areas. Single-photon emission tomography (SPECT) may be performed in conjunction with planar bone scintigraphy to characterize uptake at the primary tumor site and when there is suspicion for lung metastases.
26Numerous other imaging techniques and modalities are being used or developed for use in the assessment of osteosarcoma. Thallium-201 has shown value in evaluating tumor response to chemotherapy, but has not gained widespread application. Similarly, DCE-MRI, which provides information on tissue vascularization and perfusion, capillary permeability, and the interstitial spaces, has shown promise for the assessment of tumor response and disease prognosis.
27 Diffusion-weighted MRI sequences, which also can be performed in conjunction with standard MRI, are also being investigated for the assessment of tumor response.
28 In addition, whole-body MRI has shown potential as a replacement for bone scintigraphy for the evaluation of bone metastases.
29 Presently, however, FDG-PET and PET/CT seem to have the most potential for widespread application. FDG-PET is equal or worse than MDP-bone scintigraphy for the detection of bone metastases and worse than CT for the detection of lung metastases.
30 FDG-PET, however, has shown promise in the assessment of tumor response and the detection of recurrent disease.
31,32As imaging techniques evolve and new techniques are developed, the basic imaging assessment for children with osteosarcoma will continue to rely on conventional radiography, MRI, CT, and bone scintigraphy. There is general agreement on the need for
MRI of the primary tumor at presentation and prior to local control and the use of bone scintigraphy and chest CT for the assessment of metastatic disease at presentation.
3,33 There are differing opinions regarding the timing and types of imaging that should be performed later in the patients’ treatment course and during surveillance posttherapy.
3,20,33 These differences will likely continue until there is objective evidence on the value of a standard protocol of imaging studies to improve the outcome of children with osteosarcoma.
Classification and Staging
Tumor extent (local and systemic) as well as the grade of the tumor must be taken into account when classifying and staging bone tumors. For many years, clinicians, particularly orthopedic surgeons, have relied on a staging system developed initially by Enneking and subsequently revised by the Musculoskeletal Tumor Society (MSTS), which recognizes these requirements. In the MSTS system, tumors are classified as either intra- (T1) or extracompartimental (T2) and as either low-grade (A) or high-grade (B) malignancies. Metastatic tumors are classified as T3.
34 Since extension through the cortex to the periosteum (present in virtually all osteosarcomas) defines extracompartimental involvement, the MSTS staging system classifies the majority of localized pediatric and adolescent osteosarcomas as stage IIB (
Table 34.2). The T-stages of the current 6th edition of the UICC TNM classification of malignant tumors,
35 an advancement over the MSTS staging system, distinguishes between smaller and larger primary tumors, with a cutoff at 8 cm in the greatest dimension, and also allows for the description of skip metastases as T3 (see
Table 34.3). The 6th edition of the UICC TNM classification also allows the distinction between pulmonary (M1a) and extrapulmonary (M1b) distant metastases. The resulting staging system is reminiscent of the original MSTS classification in that both grades of malignancy and local tumor extent are used for definitions.
35Regardless of the staging system, the vast majority of children and adolescents diagnosed with osteosarcoma will fall into two groups. First, those with high-grade osteosarcoma with tumor radiographically evident at the primary site and second, those with tumor radiographically visible at metastatic sites. Although the presence of radiographically visible metastatic disease has a major influence on prognosis, the chemotherapy typically used is similar. Indeed, virtually all patients with high-grade osteosarcoma and adequate organ function are treated with similar chemotherapy regimens which will be discussed further subsequently.
Biopsy
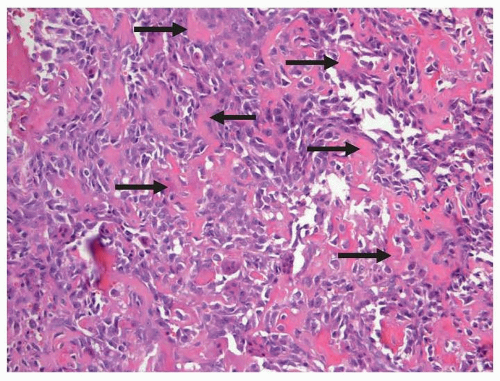
Although the index of suspicion of osteosarcoma can be exceedingly high based on the radiograph alone, a pathologic confirmation of that diagnosis is required. In other cases, the radiographic appearance may not be typical of osteosarcoma and a benign lesion may be suspected. As the routine management of osteosarcoma has evolved so that the resection of the entire tumor is typically performed after a period of neoadjuvant or induction chemotherapy, the initial surgical procedure will typically be an incisional biopsy, just acquiring a sample for diagnostic purposes. Incisional biopsies can be performed in different manners and include needle (closed) and open biopsies. Needle biopsies can be either fine needle or core. Indeed, the topic of whether the biopsy should be performed as a closed or open biopsy is immensely controversial. The clear advantage of a needle biopsy is that it can be performed in an interventional radiology suite under radiographic guidance or in an office setting with limited sedation, with prompt patient recovery, and at a reduced cost. The use of local anesthetic in children and adolescents for the purposes of a biopsy generally is not adequate. The clear advantage of an open biopsy is that much more tissue can readily be obtained. The debate as to what technique is preferred encompasses issues as to who performs the procedure in each setting, issues of operating room and hospital bed availability, patient discomfort and convenience, and defining how much tissue is needed to make the diagnosis.
At one end of the spectrum, fine-needle biopsies are often the easiest procedure to perform but obtain the least tissue. Although some reports suggest that the accuracy of a fine-needle aspiration at diagnosing osteosarcoma may be as high as 90% in the hands of an experienced cytopathologist, it must be recognized that the radiograph alone in some osteosarcoma cases may have a similar specificity. Coupling classic radiographic findings to the presence of a malignant spindle cell on cytopathology may yield in aggregate sufficient diagnostic information.
Core biopsies allow visualization of the architecture of the tumor and tissue providing much more diagnostic material particularly when multiple cores are obtained. A risk of a core biopsy is that it enters the tumor deeply and can lead to bleeding and tumor contamination. Taking multiple cores provides additional tissue, but each additional core has a risk of compromising the area’s structural integrity. Needle biopsies, either fine needle or core, can also lead to a diagnostic delay. Permanent sections which may take several days to process can ultimately result in a specimen in which the diagnosis is indeterminate, which can occur in 25% to 30% of cases, even at experienced centers.
Open biopsies yield the most tissue and when performed by an experienced orthopedic surgeon, rarely cause complications. The debate on how much tissue is needed relates to the fact that the diagnosis of osteosarcoma is based on identifying a region of the tumor with the defining histologic appearance. Molecular analysis is typically used only to exclude other diagnoses as will be described further subsequently in the section on Pathology. Unlike other pediatric malignancies such as leukemia and neuroblastoma, biologic features of the tumors do not presently define prognosis or treatment.
Excisional biopsies are rarely performed in osteosarcoma as most of these tumors are large at presentation and not easily amenable to removal and reconstruction without significant planning. These types of extensive operations are usually not undertaken without establishing the biologic nature of the entity being removed as this heavily influences the desired surgical margins. It should be recognized that although induction chemotherapy is typical, it does not produce a survival advantage.
36 In cases in which multiple biopsies obtaining large amounts of tissue do not yield a diagnosis, an excisional biopsy or pretreatment resection could be appropriate. In these cases, the surgical approaches and margins would need to appropriate for resection of an osteosarcoma. Curettage approaches used for benign bone lesions do not yield adequate surgical margins and potentially disseminate tumor locally. This can occur if a biopsy is not performed prior to excision in a lesion that is believed to be benign based on radiographic appearance and is discovered to be an osteosarcoma.
Regardless of the type of biopsy, its placement relative to the location of the tumor and the anatomic structures of the patient are also of critical importance. The location of the biopsy site is determined by a thorough prebiopsy assessment of the extent of local disease and its relationship to critical structures such as the neurovascular bundle. This must be determined on a case-by-case basis. It is strongly recommended that the biopsy be performed by the surgeon or at least by a team that is familiar with the individual who will be performing the definitive resection so that the biopsy tract can be ellipsed within the planned surgical incision. At the time of biopsy, the individual performing it must be familiar with orthopedic oncologic principles of flap development, coverage and even amputation, when definitive limb salvage is the proposed plan for a given bone tumor. Thus, the critical message is that a biopsy needs to be performed to make the diagnosis of osteosarcoma and this biopsy needs to be performed by an individual who is experienced in the diagnosis and surgical management of this entity.