5.1
Introduction
A unique feature of bone is that it contains specialized cells dedicated to its own destruction. These cells, the osteoclasts, are generally regarded as the only cells capable of resorbing bone. Osteoclasts are motile, usually multinucleated cells, derived from promonocytic precursors present in marrow, spleen, and circulating blood. They are considered to be terminally differentiated cells that are incapable of self-replication. Osteoclasts excavate bone and other mineralized tissues by secreting acid and a proteolytic enzyme (cathepsin K) into a sealed extracellular compartment to dissolve the mineral and collagenous matrix components, creating characteristic pits and troughs with sharply defined edges ( Figs. 5.1 and 5.2 ).
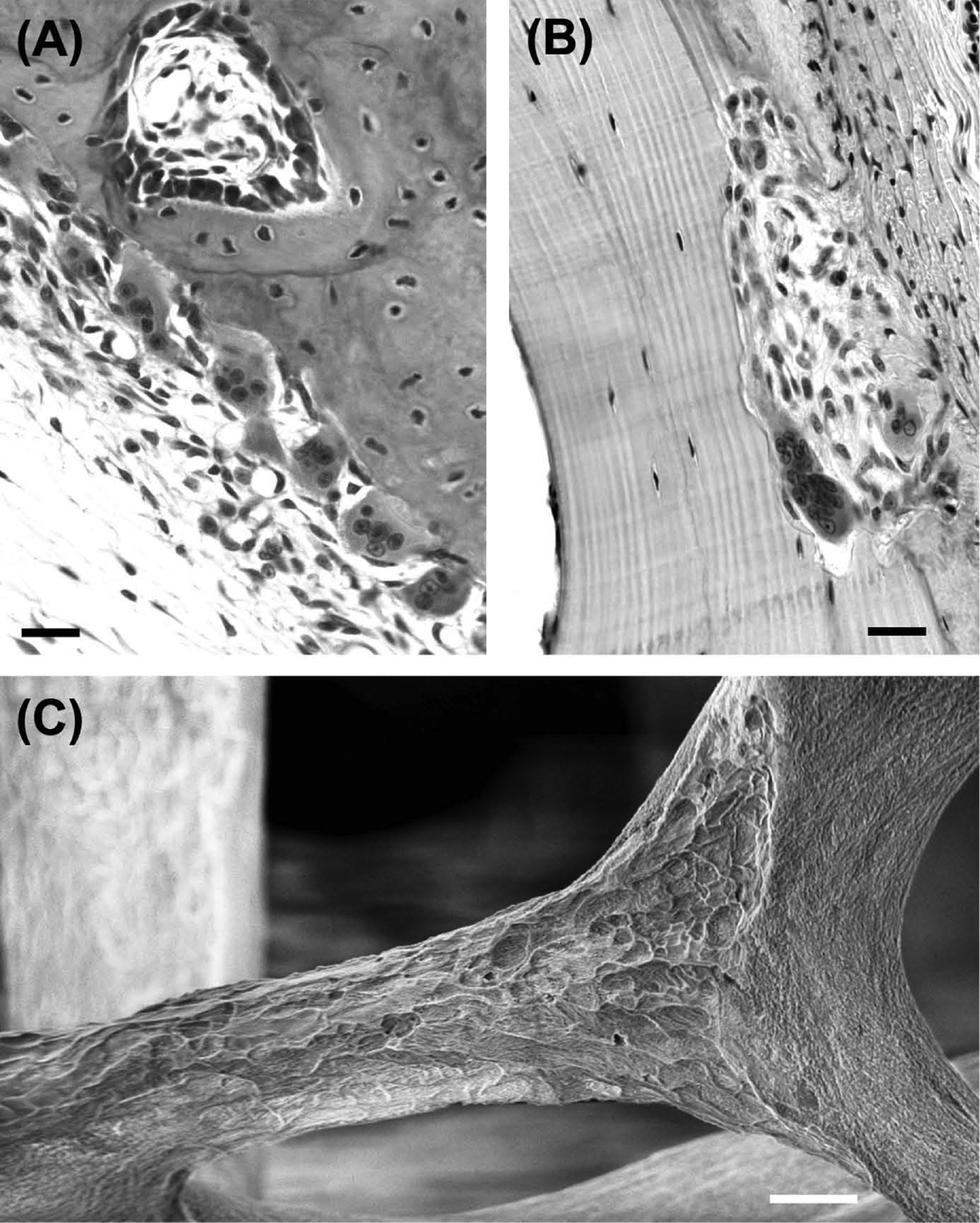
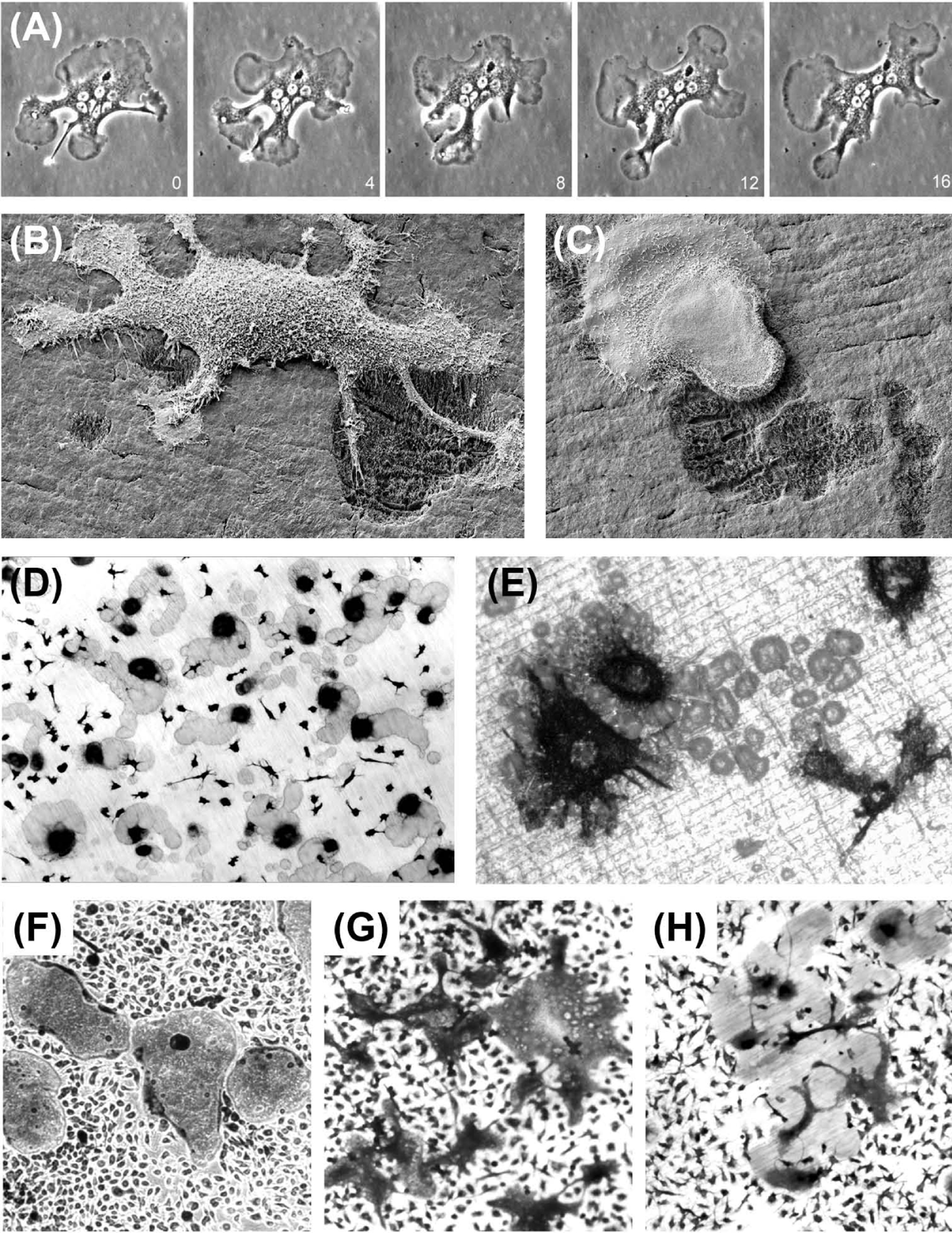
With the wider appreciation of the impact of common bone loss disorders, the years since the 1980s have seen a great increase in research on the osteoclast. This has led to critical advances in our understanding of the biology of these remarkable cells, and the development of effective antiresorptive treatments.
The molecular pathways involved in the regulation of osteoclast formation and function have been extensively reviewed in a numerous, excellent articles . This review aims to provide an additional focus on aspects of osteoclast physiology and function that have been less well-covered of late.
5.2
Regulation of osteoclast formation
5.2.1
Early research
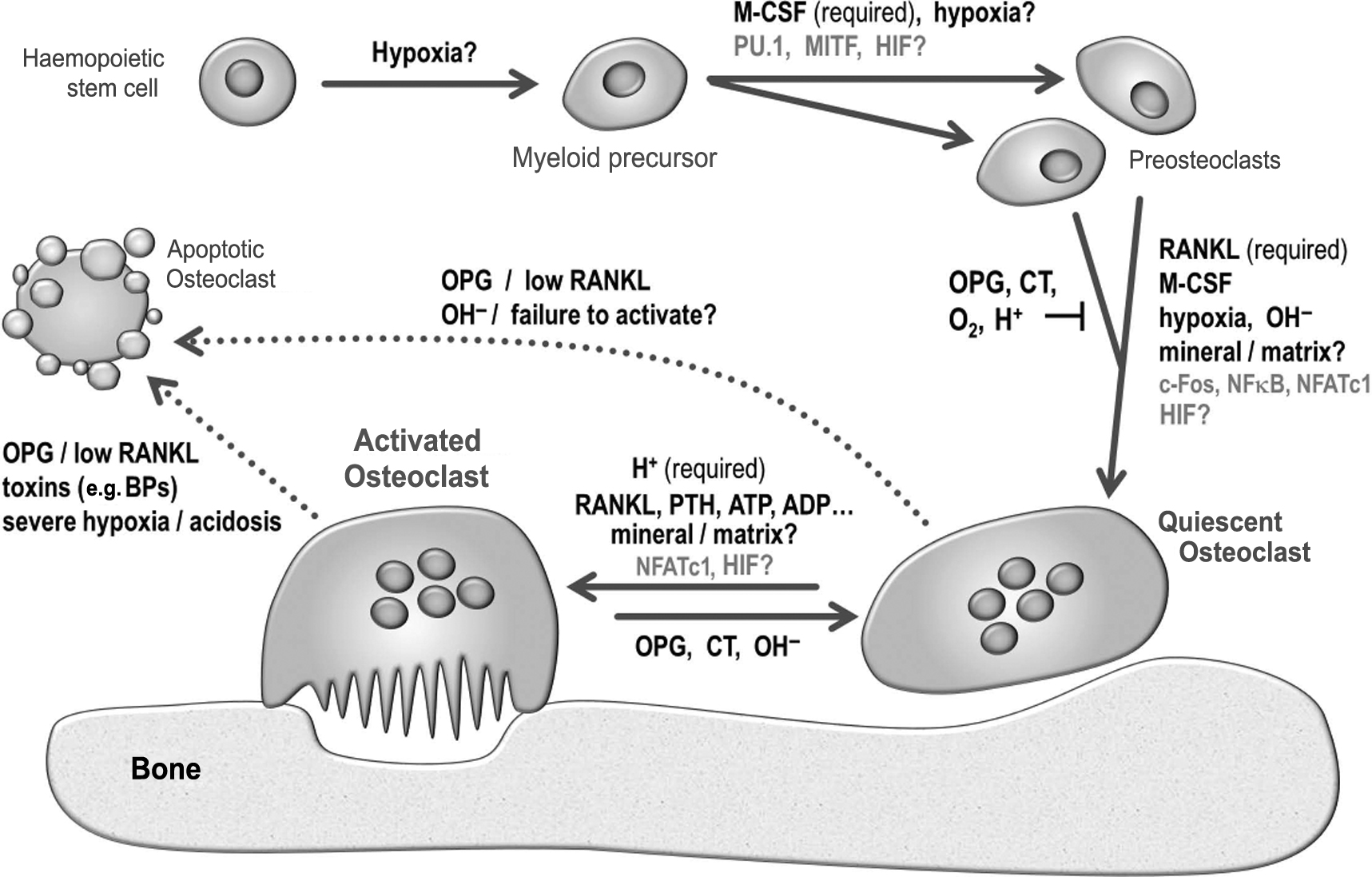
Osteoclasts were first described and named by Kölliker in 1873 , although resorption pits had been observed by Howship in the 1820s. The prevailing view for much of the next century after their discovery was that osteoclasts originated from an osteoblastic or chondrocytic precursor. Hancox was the first to propose, in 1946, that osteoclasts derived from the fusion of monocytic precursors . The hematopoietic origin of osteoclasts was firmly established in the 1970s by Walker, who showed that temporary parabiosis and grafts of marrow or spleen cells could restore bone resorption and cure osteopetrosis in the gray-lethal mutant mouse strain . The next major advance in understanding osteoclast formation came from the study of another osteopetrotic mouse strain, op/op , which also exhibited severe reductions in circulating monocytes and peritoneal macrophages . The op/op mice were shown to harbor an inactivating mutation in the gene encoding production of macrophage colony-stimulating factor (M-CSF) by fibroblasts . Administration of exogenous M-CSF to op/op mice restored osteoclastic function, leading to the formation of marrow cavities, and thus curing the osteopetrosis .
5.2.2
Intracellular and transmembrane factors
A number of intracellular protein factors that play key roles in controlling osteoclast formation were identified from the 1990s onward. It was shown that mice deficient in the PU.1 transcription factor failed to produce macrophages and osteoclasts, exhibited the classic signs of osteopetrosis, and could be cured by marrow transplantation . Hemopoietic cells from PU.1 mice also failed to produce monocytes and lacked M-CSF receptors . These results demonstrated that PU.1 regulates osteoclastogenesis via early effects on myeloid differentiation. Deficiencies in the microphthalmia-associated transcription factor (MITF) were also found to result in osteopetrosis in mice but macrophage production still occurred; MITF was shown to increase expression of the osteoclast-macrophage marker tartrate-resistant acid phosphatase (TRAP), indicating that it regulates osteoclast formation at a later stage in the myeloid differentiation pathway than PU.1 . Deletion of the gene encoding the early response transcription factor Fos was also found to block osteoclast (but not macrophage) formation, resulting in osteopetrosis , whereas the Fos-related protein, Fra-2, appeared to be a negative regulator of fetal osteoclastogenesis . Also required for the formation of multinucleated osteoclasts are the osteoclast and dendritic cell-specific transmembrane proteins, OC-STAMP and DC-STAMP . Notch signaling, a widely conserved system for communication between adjacent cells that uses four different transmembrane receptor proteins and five ligands, has been recognized to play key roles in bone. Notch 1 and Notch 2 appear to play significant roles in inhibiting or inducing osteoclastogenesis, respectively .
5.2.3
RANKL-RANKL-OPG axis
The discovery in the late 1990s of RANKL [receptor activator of nuclear factor kappa B (NFκB) ligand], a new member of the TNF (tumor necrosis factor) cytokine family, was a major breakthrough in osteoclast biology. RANKL was identified following the initial discovery of its circulating “decoy” receptor, osteoprotegerin (OPG), overexpression of which completely blocked bone resorption in vivo, causing profound osteopetrosis . RANKL (also independently studied as osteoclast differentiation factor, and TNF-related activation-induced cytokine ) was subsequently shown to be both necessary and sufficient for osteoclast formation from promonocytic precursor cells. Overexpression of RANKL causes massive bone resorption, whereas RANKL deficiency causes severe osteopetrosis and failure of tooth eruption. RANKL is a 27 kDa protein that exists in both membrane-bound and soluble forms. It acts on osteoclast precursor cells via its receptor, RANK and the TNF receptor–associated factor proteins TRAF2, TRAF5, and TRAF6 to activate the transcription factor, NFκB, which, in turn, activates the transcription factor, nuclear factor of activated T cells (NFATc1, deficiency of which also causes severe osteopetrosis). RANKL and OPG are produced by cells of the osteoblast lineage; the relative contributions of each cell type (precursor cells, active/quiescent osteoblasts, and osteocytes) to the production of these critical regulators is still not entirely clear . The availability of high-potency, biosynthetic RANKL (and M-CSF) has revolutionized the study of osteoclasts by enabling their generation in relatively large numbers from cultures of marrow, spleen, and peripheral blood mononuclear cells. Readers are referred to the many excellent review articles on the RANK–RANKL–OPG signaling axis .
5.2.4
Other local and systemic factors
Many other cytokines and growth factors have also been reported to stimulate osteoclastogenesis, including TNFα, granulocyte-M-CSF, interleukins 1, 6, 12, 17, 18, and 23, and transforming growth factor β (TGFβ) . Other hormonal or paracrine agents such as parathyroid hormone (PTH) , 1,25(OH) 2 vitamin D 3 , and adenosine triphosphate (ATP)/adenosine diphosphate (ADP) are also stimulatory. All these factors are likely to function either by increasing production of RANKL from osteoblasts and related cells or by acting directly on osteoclast precursors in the presence of RANKL to potentiate osteoclast differentiation.
Hormonal and paracrine inhibitors of osteoclast formation (other than OPG) include calcitonin, calcitonin gene–related peptide, amylin , estrogen , leukemia inhibitory factor , interleukins 4, 10, 13, and interferon γ . Wnt ligands promote bone formation and exert both indirect inhibitory and stimulatory actions on osteoclastogenesis . Sclerostin, the bone-specific inhibitor of Wnt signaling, is reported to stimulate osteoclast formation and resorption in cocultures with MLO-Y4 osteocyte-like cells, but to have no direct action on osteoclast precursors . In vivo, antisclerostin antibody treatment exerts a significant antiresorptive action . At a more fundamental level, acidosis exerts striking effects on osteoclast multinucleation in the presence of M-CSF and RANKL. In contrast with studies reporting increased size of osteoclasts formed from cultured mononuclear precursors , results from our own laboratory showed that acid-activation ( see the next section ) halts cell fusion, leading to increased numbers of smaller osteoclasts in mouse marrow cultures ( Fig. 5.2G,H ). Another overlooked environmental factor affecting osteoclastogenesis is temperature: intriguingly, mouse osteoclast formation and resorption pit formation were shown to be moderately enhanced in cultures incubated at 35.5°C or 34°C, compared with 37°C, whereas osteoblast function was markedly inhibited. Although the wider significance of these observations remains unclear, it is noteworthy that core body temperatures are often reduced (<35.5°C) in elderly humans .
5.2.5
Hypoxia
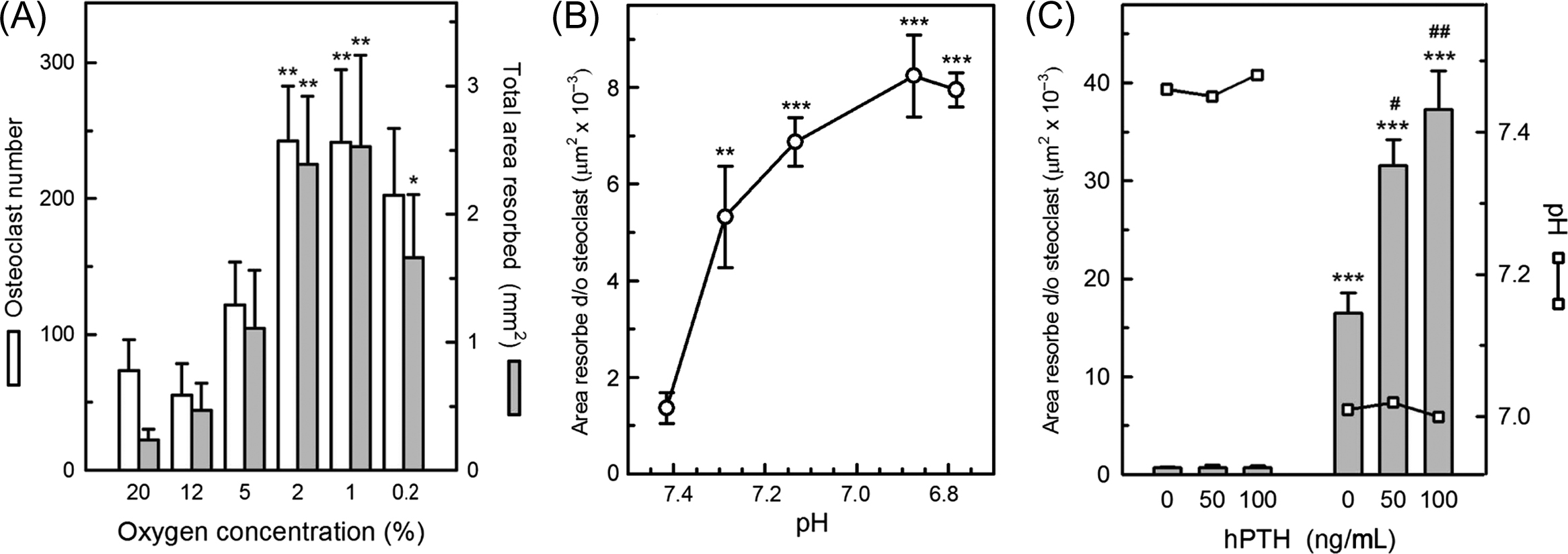
Oxygen availability is an important fundamental regulator of osteoclast formation. In the presence of M-CSF and RANKL, reducing oxygen tension from atmospheric levels (with brief reoxygenation every 2–3 days) causes striking stimulation of the number and size of osteoclasts formed in cultures of marrow or peripheral blood-derived mononuclear precursor cells, leading to large increases in resorption pit formation, peaking around 1%–2% O 2 ( Fig. 5.5A ) . In complete contrast, osteoblast differentiation and bone formation in vitro are blocked by hypoxia .
These responses need to be considered in the context of the oxygen tensions likely to be experienced by osteoclasts or their precursors in vivo. In arterial blood the pO 2 value is normally about 12% and in venous blood it is about 5%. Mean pO 2 values of 6.6% and 9.4% have been measured in normal bone marrow aspirates and by needle probe within healthy human mandible, respectively . Direct imaging measurements in the calvariae of live, young adult mice indicated a pO 2 of 6.3% in periosteum and the surprisingly low value of <2.5% in bone marrow . In pathological states, oxygen levels in bone may be particularly low. For example, pO 2 was measured at 3.7% in osteomyelitic human mandible and 0.8% in fracture hematomas of rabbit fibula . An important corollary of the previous observations is that in vitro investigations of osteoclast function and metabolism performed in atmospheric oxygen (pO 2 =21%) are actually conducted in significantly hyperoxic (thus inhibitory) conditions that constitute a 3–10-fold excess of oxygen, with respect to the physiological environment in bone. Intermittent severe hyperoxia (95%–100% O 2 ) is reported to cause further inhibition of osteoclastogenesis, in comparison with atmospheric pO 2 .
The hypoxia inducible factors (HIFs) are key intracellular transcription factors that mediate cell responses to hypoxia. Mice deficient in the Fos-related transcription factor, Fra-2 were reported to have hypoxic bones that express elevated levels of HIF-1α and contain giant osteoclasts, suggesting that HIF could play a role in osteoclast formation . However, more recent work indicated that HIF-1α does not exert a significant regulatory action on human osteoclast differentiation (despite enhancing resorption) .
The trophic response of osteoclast precursors to hypoxia probably reflects their myeloid origin: the formation or function of other cell types in this lineage is also known to be stimulated by hypoxia . These findings help one to explain why extravasation of mononuclear precursors into relatively oxygen-deficient bone microenvironments could result in osteoclast formation and suggest a new mechanism for the bone loss associated with the numerous pathophysiological conditions where tissue hypoxia may occur , such as inflammation, fractures, unloading, infection, tumors, diabetes, smoking, obstructive pulmonary disease, and anemias.
5.3
Osteoclast function
For resorption to occur, osteoclasts must be recruited to specific remodeling sites, attach tightly to exposed bone surfaces, then polarize and activate as they deploy the specialized machinery of resorption. At the end of a resorption phase, osteoclasts may either move to another location or die. Key aspects of these processes are discussed next.
5.3.1
Recruitment
Osteoclasts are normally multinucleated, although significant variation exists. For example, resorptive osteoclasts formed in vitro from human peripheral blood mononuclear cells tend to have fewer than four nuclei, including a significant number of mononuclear osteoclasts ( Fig. 5.2D,E ), whereas osteoclasts formed from mouse ( Fig. 5.2G ) or cat mononuclear cells can be very large indeed. Striking differences in morphology are seen in osteoclasts formed within a single culture from marrow (or isolated freshly from fragmented bones) ( Fig. 5.2A–C,G,H ).
Site-specific recruitment of osteoclasts from resident or circulating progenitor cells could occur via local production of RANKL and M-CSF, plus other potentiating cytokines or growth factors, as well as in response to localized hypoxia (see previous section). Mature, fully differentiated osteoclasts are also highly motile ( Fig. 5.2 ) and capable of moving rapidly to new locations. The movement of osteoclasts or their precursors along chemotactic gradients remains poorly studied, however . To perform their resorptive function, osteoclasts must be able to gain access to the exposed surfaces of bone and other mineralized tissues. This process could be facilitated by the movements or shape changes of the osteoblastic lining cells that normally cover bone surfaces, for example, in response to stimuli such as PTH .
Although the structural component of the organic matrix of bone is insoluble and normally protected by osteoblastic cells, soluble signaling proteins such as members of the TGFβ family could be released from the matrix, either during osteoclastic resorption , or perhaps even via fluid flow through osteocyte canaliculae, which have an enormous surface area of contact with bone matrix. Local damage may also play a role in recruiting osteoclasts to regions of bone requiring remodeling . For example, the density of microcracks induced by fatigue loading in rat bone correlates with activation of resorption . The mechanisms underlying such targeted remodeling are far from clear but it has been proposed that apoptotic bodies from dying osteocytes and osteoblasts could promote osteoclastogenesis . There appear to be no clear differences, though, between the susceptibility to resorption of living and devitalized bone in vitro .
5.3.2
Attachment, polarization, and resorption
Osteoclast attachment to bone surfaces is mediated mainly by the highly expressed αvβ3 integrin (vitronectin receptor). The integrin forms part of complex, dynamic adhesion structures known as podosomes that also contain actin filaments, cortactin, Wiskott–Aldrich syndrome proteins, paxillin, CD44, and attachment-related proteins such as vinculin and talin . On mineral-containing substrates, in conjunction with suitable activation signals (see section below), the podosomes become organized into a dense ring known as the sealing zone that anchors the osteoclast tightly to its substrate and isolates a compartment beneath the cell where bone destruction takes place. Podosome formation is controlled by signaling pathways involving the tyrosine kinase, c-Src, and GTPases of the Rho family . Src-deficient mice show an osteopetrotic phenotype that appears to be due to impaired osteoclast podosome formation and turnover . The Rho GTPases are key regulators of the actin cytoskeleton and motility of osteoclasts .
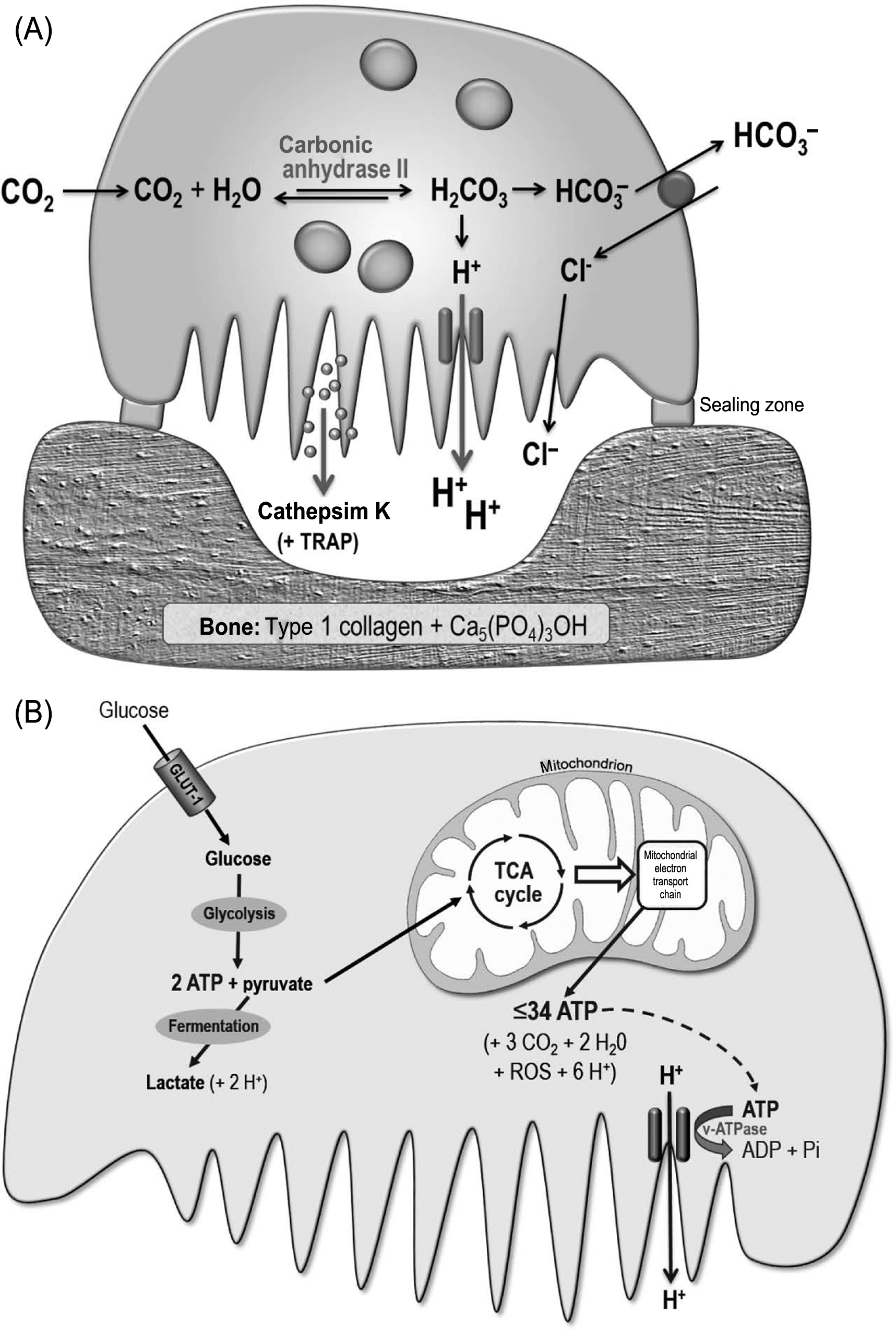
Inside the sealed resorption compartment, the cell membrane of the osteoclast develops the ruffled border, a zone of highly convoluted folds that permit a high surface area of interaction with the bone surface. Residing in the membrane of the ruffled border is the vacuolar-type H + ATPase, which actively pumps protons from the interior of the osteoclast into the sealed resorption compartment adjacent to the bone surface . The protons secreted by the v-type H + ATPase are thought to be derived mainly from carbonic acid, the cytosolic formation of which is catalyzed by carbonic anhydrase II; the residual bicarbonate ions are passively exchanged for chloride at the upper (basolateral) surface of the osteoclast . Also located in the ruffled border is the ClC-7 chloride channel that allows the passive movement of chloride ions into the resorption space; this channel is also reported to mediate Cl − /H + exchange , and its deletion results in severe osteopetrosis . The acid environment in the sealed resorption compartment serves to dissolve the bone mineral, which consists mainly of hydroxyapatite [Ca 5 (PO 4 ) 3 OH], the solubility of which is highly pH-dependent. The acid secreted by the osteoclast is continuously neutralized by the hydroxyl moiety of the hydroxyapatite mineral. It has been proposed that pain may result if the acidic resorption compartments expose sensory nerve fibers present within living bone—for example, during the aggressive resorption associated with some cancers .
The degradation of the acid-demineralized organic component of the bone matrix, which is about 90% type 1 collagen, is primarily due to the action of cathepsin K, a member of the papain family of cysteine proteases that is highly (although not exclusively) expressed by activated osteoclasts and secreted in vesicles across the ruffled border. Cathepsin K readily degrades type 1 collagen, with optimal activity at about pH 6.0; it cleaves all the three chains of the collagen triple helix, as well as the telopeptides, generating a variety of peptide fragments in the 70–80 kDa weight range. The overall effectiveness of cathepsin K in solubilizing collagen is similar to that of bacterial collagenase. Loss-of-function mutations in the cathepsin K gene are responsible for the rare human disorder pycnodysostosis, characterized by osteosclerosis, short stature, and skeletal malformations. Mice deficient in cathepsin K show increased trabecular and cortical bone mass due to impaired bone resorption . The large collagen fragments generated by the action of cathepsin K are thought to be endocytosed across the ruffled border of the osteoclast, trafficked across the cell cytoplasm, and exocytosed across the upper cell membrane into the extracellular environment , where they are presumably subject to further degradation by other enzymes, including metalloproteinases. This process may help one to account for the very rapid degradation of bone matrix achieved by osteoclasts (see next). Osteoclasts also highly express TRAP, an enzyme that serves as a convenient marker for osteoclasts, although its functions are still not well-understood . TRAP generates reactive oxygen species that may aid matrix degradation but does not appear to play a critical role in bone resorption, since TRAP-deficient mice show only mild osteopetrosis .
The sealed resorption compartment may be considered as an extracellular lysosome. The concentrated action of acid and cathepsin K within this space results in the creation of sharply demarcated resorption pits and troughs, which frequently show exposed, demineralized collagen fibers at their base. The resorption process is facilitated not only by the motility of the osteoclast ruffled border but also by the migration of the whole cell, resulting either in groups of discrete pits, continuous trails or larger, conjoined areas of excavation ( Fig. 5.2 ). The key processes involved in osteoclastic resorption are summarized in Fig. 5.4 .
5.3.3
Osteoclast activation
The activation of osteoclasts involves upregulation of the critical cellular machinery needed for resorption pit formation, chiefly the sealing zone/podosomes and ruffled border, the v-type H + ATPase, the ClC-7 channel, carbonic anhydrase II, and cathepsin K.
Although many local and systemic factors can stimulate osteoclast function in intact bone, cell culture studies indicate that only one extracellular agent, hydrogen ions (i.e., acidity), can activate osteoclasts directly to form resorption pits. Mature rat osteoclasts were found to be almost inactive at pH 7.4 (corresponding to “physiological” or blood pH) but showed steeply increased resorption pit formation as pH was reduced, reaching a plateau at about pH 6.8 . Subsequent studies showed that avian and human osteoclasts also exhibit acid-activation responses. The sensitivity of osteoclasts to extracellular H + is such that pH reductions of <0.1 U can cause a doubling of resorptive activity ( Fig. 5.5B ). Osteoclast-mediated calcium release from intact bone in organ culture also shows impressive sensitivity to acid stimulation .
Extracellular acidification rapidly stimulates the formation of sealing zones/podosomes and the expression of carbonic anhydrase II, the v-type H + ATPase, cathepsin K, TRAP, and TRAF6 in osteoclasts . Lowering extracellular pH also greatly increases nuclear accumulation (activation) of the key transcription factor NFATc1 to levels comparable with those induced by RANKL; unlike RANKL, however, acidosis does not appear to induce nuclear translocation of NFκB. Both acidosis and RANKL additionally cause transient elevation of cytosolic-free Ca 2+ in osteoclasts . The mechanisms by which osteoclasts sense extracellular pH are still unclear and are likely to be complex, potentially involving both pH-sensitive ion channels and cell surface receptors, such as ASICs 1–3 and OGR1 , as well as direct intracellular responses to changes in cytoplasmic pH .
Many of the potential causes of systemic or local acidosis are associated with bone loss (e.g., renal and respiratory disease, gastroenteritis, anaerobic exercise, diabetes, aging, the menopause/androgen deficiency, consumption of acidifying substances, ischemia/hypoxia, inflammation, fractures, and tumors ). The requirement of osteoclasts for acid-activation may be related to their white blood cell lineage. Dendritic cells that share a myeloid origin with osteoclasts also show major activation responses to low pH . The activation of osteoclasts by acid could be considered as a primitive homeostatic response to correct systemic acidosis by increasing the release of alkaline bone mineral (hydroxyapatite).
As described in a preceding section, hypoxia stimulates the formation and multinucleation of osteoclasts. However, the effect of hypoxia on the resorptive activity of osteoclasts remains somewhat controversial. Experiments with freshly isolated mature rat osteoclasts from rat bones showed clearly that in pH-controlled conditions, hypoxia did not alter their resorptive activity . In non-pH-controlled cultures, hypoxia (which causes acidosis) is reported to stimulate the resorptive activity of human osteoclasts . A more recent study suggested that HIF-1α enhances osteoclast activity via the oxygen-sensing enzyme, prolyl-4-hydroxylase 2 .
Once activated by acidosis, osteoclasts can be further stimulated by factors such RANKL , PTH ( Fig. 5.5C ), 1,25(OH) 2 vitamin D , and ATP/ADP . It is worth noting that RANKL is almost inactive on mature osteoclasts before acid-activation ; an earlier report of osteoclast activation by RANKL was based on experiments that were performed in acidified conditions . Osteoclast activation may therefore be considered a two-step process, with acidification as the obligatory, initial “on switch.”
The previous results also indicate that the stimulation of osteoclasts by PTH is not necessarily indirect (i.e., mediated via PTH actions on cells of the osteoblast lineage), as proposed by the well-known hypothesis of Rodan and Martin . Moreover, mature human osteoclasts express PTH type 1 receptors, and PTH stimulates resorption pit formation by purified populations of these cells .
The work rate of fully activated osteoclasts can be very high. In vitro, continuous resorption trails are commonly observed when osteoclasts are formed on bone or dentine from mononuclear blood precursor cells using M-CSF and RANKL, then acid-activated. For the relatively small human osteoclasts illustrated in Fig. 5.2D , the resorption trails formed over 2 days are about 150 µm long, 50 µm wide, and are typically about 10 µm deep. Thus each osteoclast removed about 30,000 µm 3 of mineralized matrix per day, equivalent to about twice its own volume. These observations emphasize the requirement for tight control of osteoclast function in vivo.
The major physiological inhibitor of osteoclast function appears to be OPG . Mammalian osteoclasts express abundant receptors for the inhibitory peptide hormone calcitonin and respond to calcitonin with a rapid cessation of motility and resorptive function but calcitonin does not seem to be a significant regulator of osteoclast function in normal adults in vivo .
5.3.4
Osteoclast survival
The key factor affecting osteoclast survival is probably the balance between the RANKL and OPG levels to which the cell is exposed. Withdrawal of RANKL or increased OPG strongly promotes osteoclast apoptosis . Many other pathophysiological factors can potentially reduce osteoclast lifespan, including extended hypoxia , glucocorticoids , severe acidosis , and high concentrations of extracellular ATP . Mild acidosis, on the other hand, is reported to exert a protective effect on osteoclast survival, mediated through an NFAT-independent, protein kinase C-dependent pathway . Unpublished work from our laboratory indicates that the survival of mouse osteoclasts formed from marrow precursors using M-CSF and RANKL is impaired if cultures are maintained at “physiological” pH (7.4) for extended periods, so that they cannot become activated to excavate resorption pits .
The impact of hypoxia on osteoclast survival depends on its severity and duration. Exposing cultures of freshly isolated, mature rat osteoclasts to hypoxia (1%–2% of O 2 ) or near-anoxia (0.2% of O 2 ) for 26 hours reduced their numbers by 50% and 75%, respectively . This observation suggests that the large increases in osteoclast numbers and resorption seen in the longer term osteoclast-forming mouse marrow and human peripheral blood mononuclear cell (PBMC) cultures in very low O 2 (but with intermittent reoxygenation) may have occurred against a background of increased cell death. Independent studies showed that culturing osteoclast-forming human PBMCs in continuous hypoxia (2% of O 2 ) without reoxygenation for periods up to 3 days led to net reductions in osteoclast numbers . Fluctuating, nonuniform oxygen levels may more accurately reflect the in vivo microenvironments experienced by differentiating osteoclasts than steady state hypoxia .
The process of resorption itself may also adversely affect osteoclast survival by increasing exposure of the cell to toxins released from bone matrix. An obvious example is the induction of osteoclast apoptosis following endocytosis of bisphosphonates adsorbed to bone surfaces . It is also possible that a component of the beneficial skeletal effects reported for strontium ranelate therapy could be due to increased osteoclast apoptosis following ingestion of strontium ions incorporated into bone .
Some of the key regulatory pathways involved in osteoclast formation and function are summarized in Fig. 5.3 .
Related posts:
 The bone organ system: form and function
The bone organ system: form and function
 Impact of physical characteristics and lifestyle factors on bone density and fractures
Impact of physical characteristics and lifestyle factors on bone density and fractures
 Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
 Relationship between periodontal disease, tooth loss, and osteoporosis
Relationship between periodontal disease, tooth loss, and osteoporosis
 On the evolution and contemporary roles of bone remodeling
On the evolution and contemporary roles of bone remodeling
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


