INTRODUCTION
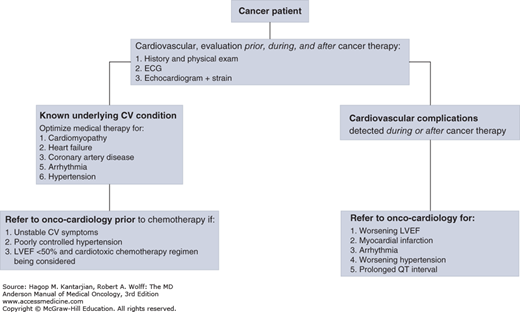
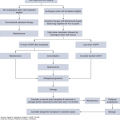
Onco-cardiology is a fast-growing medical subspecialty focused on the management of heart diseases in patients with cancer. Although cancer remains a leading cause of morbidity and mortality worldwide, the survival rate of patients with cancer has increased in the last 25 years. In the United States, the 5-year relative survival rate of patients diagnosed with cancer between 1975 and 1977 was 50%; it increased to 68% between 1999 and 2005. The US National Cancer Institute estimates that at least 13.7 million cancer survivors were alive in the United States in 2012 (1). With the survival improvement, the long-term adverse treatment effects have also become more apparent. A survey of 1,807 cancer survivors with a 7-year follow-up found that 33% died of heart diseases and 51% died of cancer (2). Historically, since the late 1970s, the interest in chemotherapy-induced cardiotoxicity was focused on cardiomyopathy related to few chemotherapeutic agents. As the field of cancer therapies has expanded, so has the finding of other cardiovascular side effects such as transient left ventricular dysfunction, hypertension (HTN), cardiac arrhythmias, pericardial effusions, and arterial ischemia. Patients with known or subclinical cardiac disease are more susceptible to the cardiotoxic side effects of cancer therapy, and those with known cardiac disease often need to alter their cardiac management to allow for the treatment of cancer. This can be associated with significant cardiovascular risks. Onco-cardiology has evolved to address the cardiovascular needs of patients whose optimal outcome mandates close and collaborative efforts between cardiologists and oncologists in a multidisciplinary approach. Involvement of cardiologists in cancer patients’ care has changed from focusing on management of the cardiovascular complications of therapy to an overall assistance in the care of these patients from the initial cancer diagnosis to survivorship as outlined in Fig. 54-1.
PATHOPHYSIOLOGY
Clinical knowledge and basic science discoveries in onco-cardiology have grown over the last decade. There is better understanding of molecular mechanisms of cardiac toxicity of several cancer drugs. An example is the observation that the Bruton tyrosine kinase receptor is expressed in the atria of human heart tissue (3). This tyrosine kinase receptor is the target of a novel drug, ibrutinib, used in certain hematologic malignancies including chronic lymphocytic leukemia. Ibrutinib has been associated with a significant incidence of atrial arrhythmias, potentially mediated by its on-target effect and more specifically by inhibiting the PI3K-Akt signaling pathway (3). Similarly, trastuzumab targets ErbB2 in breast cancer cells and has been shown to improve outcome and prolong survival in HER2+/ErbB2 breast cancer. The same receptors within the cardiac myocyte are responsible for normal cell function and myocyte repair. When affected by ErbB2 inhibitors like trastuzumab, secondary mitochondrial dysfunction can be observed (4). Other mechanisms of toxicities have recently been reported with older conventional chemotherapeutic drugs like the anthracyclines; cardiovascular toxicity of this drug has been recently linked to inhibition of topoisomerase IIβ (5). This enzyme is responsible for normal myocyte function, and its inhibition partially explains the cardiovascular toxicity of doxorubicin. In addition to the benefit of improving strategies to prevent drug toxicities, a better understanding of these biologic pathways may also provide better understanding of certain heart diseases and their management. For example, the discovery of the key role of ErbB2 in normal cardiac function and its relation to trastuzumab toxicity was followed by the discovery of the benefit of an ErbB2 agonist called neuregulin-1 in the animal models of heart failure (6). Neuregulin-1 is currently being studied for the treatment of heart failure in humans.
CARDIOVASCULAR SYNDROMES OBSERVED IN CANCER PATIENTS
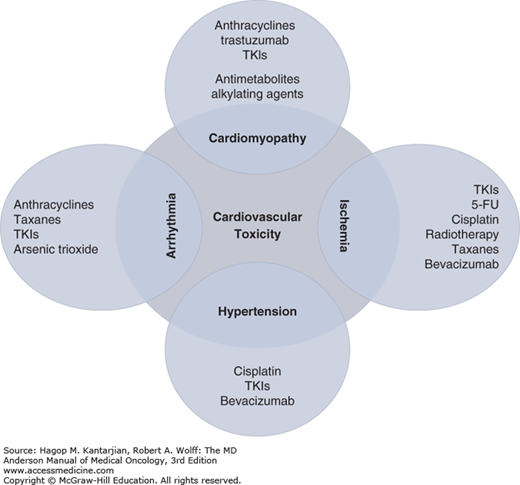
Aside from the direct cardiac toxic effect of certain cancer drugs (Fig. 54-2), many aspects of the cardiovascular system can be affected by cancer or cancer therapy (Fig. 54-3), resulting in a variety of cardiovascular syndromes.

Onco-cardiologists are often asked to assess patients with cancer and clinical heart failure (HF) symptoms. Although the most feared etiology is the progressive, permanent, and irreversible myocardial damage related to anthracycline toxicity, this in fact constitutes only a small percentage of patients with clinical HF. The majority of patients with new-onset HF in the setting of cancer and cancer therapy have their symptoms triggered by mechanisms other than anthracycline toxicity. Despite some overlap, a clinically useful approach is to divide patients into two groups based on the initial clinical presentation (Table 54-1). The first group includes patients presenting with sudden and new-onset acute HF with associated systolic dysfunction. These patients include those with cardiomyopathy related to sepsis, stress cardiomyopathy, and myocarditis (toxic or infectious). The second group includes patients presenting with subacute or chronic HF symptoms and includes those with underlying structural heart disease, those with chemotherapy-induced cardiomyopathy, and those with infiltrative myocardial conditions related to their underlying malignancies or to cancer therapy (amyloidosis, iron overload). Evaluation of these patients typically includes obtaining a two-dimensional echocardiogram, brain natriuretic peptide, troponin, thyroid function tests, iron/ferritin level, ischemia assessment, and occasionally endomyocardial biopsy.
| Acute Left Ventricular Systolic Dysfunction | Subacute/Chronic Left Ventricular Dysfunction |
|---|---|
Stress cardiomyopathy Sepsis Myocarditis Viral Chemotherapy Myocardial infarction Metabolic derangement Hypocalcemia | Preexistent cardiomyopathy Ischemic heart disease Nonischemic cardiomyopathy Hypertensive heart disease Chemotherapy Infiltrative cardiomyopathy Amyloid heart disease Iron overload |
Most of these cases are related to conditions referred to as reversible myocardial dysfunction or stress cardiomyopathy. This is a phenomenon typically triggered by sepsis, but also occurs in a wide range of acute illnesses (see Table 54-1). Myocardial dysfunction has been documented in up to 40% of patients with sepsis and is a major predictor of mortality in this population (7,8). During the course of the malignancy management, patients are at risk of these acute illnesses, especially during chemotherapy with secondary neutropenic sepsis. Physiologically, both left and right ventricular systolic functions are diminished with variable degrees of myocardial depression. This is typically reversed within 7 to 10 days. The exact mechanism for myocardial dysfunction is not well defined, and multiple theories have been suggested including the possibility of altered microcirculatory flow, mitochondrial dysfunction, myofibrillary dysfunction, autonomic dysregulation, altered calcium cellular transportation, and others (7,9) (Fig. 54-4). Clinically, patients with reversible myocardial dysfunction/stress cardiomyopathy are recognized when evidence of new-onset left ventricular (LV) dysfunction is documented in the appropriate clinical setting (see Table 54-1). It is frequently associated with repolarization abnormalities on electrocardiogram (ECG) and minimal rise in cardiac troponins. Echocardiographic findings of severe LV dysfunction are typically out of proportion to the ECG changes and the cardiac biomarkers rise. The severity of LV dysfunction, however, correlates well with the brain natriuretic peptide (BNP) and N-terminal proBNP (NT-proBNP) levels (7,9). The management is focused on stabilizing the patient’s blood pressure using vasopressors if shock is present and transitioning to cardioprotective drugs like angiotensin-converting enzyme (ACE) inhibitors and β-blockers after the patient is weaned off of pressure support and when blood pressure is stable for 24 to 48 hours. Improvement and recovery of myocardial dysfunction are typically observed within 7 to 10 days (7). Persistent LV dysfunction beyond 2 weeks should raise suspicion of possible underlying ischemic heart disease or viral myocarditis in these typically immunocompromised patients. These two conditions are associated with significant rise in cardiac biomarkers like troponin and creatine kinase (CK)-MB. Patients with suspected ischemic heart disease should undergo evaluation and management of coronary artery disease. The diagnosis of viral myocarditis is more challenging because viral cultures and antibody titers have limited diagnostic accuracy. Myocardial biopsy is the gold standard in the general population for diagnosis of viral myocarditis despite its limited diagnostic yield (60% and 80% sensitivity and specificity, respectively) (10). This can be more challenging and risky in patients with cancer because they often have coagulopathy with risk of procedure-related complications (bleeding, myocardial perforation). The use of empiric therapy with intravenous immunoglobulin for its antiviral and immunomodulatory effect in this setting is controversial, especially with the limited data to support the benefit of such intervention in the adult population (11). It is also typically associated with high-volume fluid shift, which can exacerbate HF. Figure 54-5 summarizes a suggested algorithm for evaluation of new-onset cardiomyopathy and HF in patients with cancer.
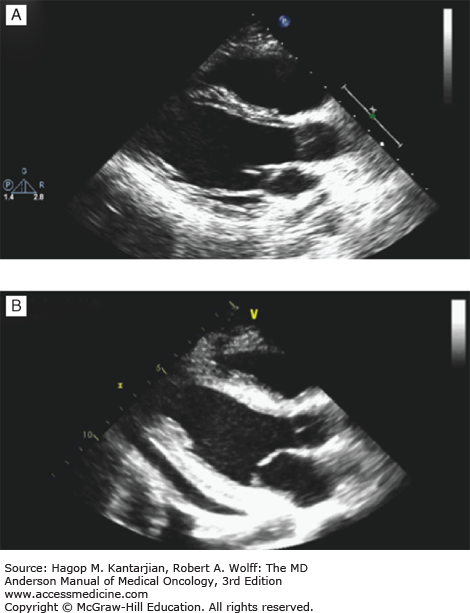
FIGURE 54-4
Acute severe cardiomyopathy with shock following chemotherapy for ovarian cancer complicated by severe hypocalcemia. Echocardiographic studies showing (A) baseline normal left ventricular systolic function and (B) severely depressed systolic function with small pericardial effusion 48 hours after developing severe hypocalcemia. (C) Severe hypocalcemia following chemotherapy.


A large number of chemotherapeutic agents have been linked to cytotoxic myocardial injury. Table 54-2 lists several groups of chemotherapeutic agents known to be associated with LV systolic dysfunction or HF. The ones most commonly associated with cardiomyopathy include anthracyclines, alkylating agents, and tyrosine kinase inhibitors (TKIs). Cardiotoxicity in general is defined as a drop in LV ejection fraction (LVEF) by 5% or more, to less than 55% in the presence of HF symptoms, or an asymptomatic drop in LVEF by 10% or more to less than 55% (12). Myocardial toxicity is also classified into type I and type II based on the nature of myocyte injury.
| Chemotherapeutic Agent | Mechanism of Toxicity | Cardiomyopathy Incidence | Other Cardiovascular Toxicity |
|---|---|---|---|
Anthracyclines
| Oxidative stress Inhibition of topoisomerase IIβ | 5% at 400 mg/m2 18% at 700 mg/m2 4% at 900 mg/m2 15% at 1 g/m2 5% | Arrhythmia Pericarditis Arrhythmia |
Anthracyclines analogues
| 2.6% | Arrhythmia Hypertension | |
Monoclonal antibodies
| No direct toxicity Hypomagnesemia-related arrhythmia Infusion-related hemodynamic effect ErbB2 receptor–mediated myocyte dysfunction VEGF inhibition Hypertension-mediated cardiomyopathy | <0.5% 2%-28% 1.7%-3% | Arrhythmia Hypotension Arterial thrombosis Hypertension |
Alkylating agents (at high dose)
| Myocarditis Myocardial fibrosis Myocarditis | 3%-25% Rare 17% | Tamponade Pericardial effusion Myocardial infarction, arrhythmia |
Antimetabolites
| Noncardiogenic pulmonary edema | 7.1% 2% | Ischemia |
Antimicrotubule agents
| Impairment of myocardial metabolism | 3% | Coronary spasm Hypertension |
Oral tyrosine kinase inhibitors
| Mitochondrial toxicity Dysregulation of cellular energy Mitochondrial toxicity | 1%-4% heart failure Up to 10%-28% left ventricular systolic dysfunction | Hypertension QT prolongation Arterial thrombosis Arrhythmia Fluid retention |
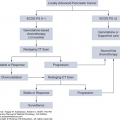
Anthracyclines are the prototype of drugs causing type I, irreversible myocyte damage. Histologic findings include myofibrillar disarray, disruption of cellular organelles, myofibrillar loss, and myocyte death (13). Myocardial toxicity is dose dependent, with <5% chance of cardiomyopathy observed at a cumulative dose of <400 mg/m2 in the case of doxorubicin (150 mg/m2 for idarubicin and 900 mg/m2 for epirubicin). This risk increases to 26% at a cumulative dose of 550 mg/m2 (14). Anthracycline-related cardiotoxicity has also been classified as acute or chronic. The acute form manifests as nonspecific ECG changes, arrhythmia, myopericarditis, and transient LV dysfunction. The more feared chronic form is marked by LV systolic dysfunction occurring many months to years following exposure and manifests as progressive HF. It is not clear whether the acute form of anthracycline cardiotoxicity is the prodrome of the more delayed cardiomyopathy form, because it has been shown that myocyte dysfunction can be observed even after the first dose of the drug. It is plausible that myocardial reserve allows normal cardiac function despite initial injury and until a second insult leads to further myocardial cell loss and subsequent systolic dysfunction. The long-term prognosis of chemotherapy-induced cardiomyopathy is much worse compared to other etiologies (15). For years, the understood mechanism has been based on the free radical and iron hypothesis. The addition of an electron to the quinone moiety of anthracyclines within the cardiomyocytes leads to the generation of excess reactive oxygen species (ROS) and subsequent mitochondrial and intracellular protein damage. This toxicity is further increased when ROS interact with iron, generating a surge of oxidative stress (16). More recently, topoisomerase IIβ was found to be a key mediator of anthracycline-induced cardiomyocyte toxicity. Anthracycline-mediated inhibition of topoisomerase IIβ causes double-stranded DNA breaks, which can lead to cardiomyocyte death (5). The use of topoisomerase IIβ level in peripheral blood leukocytes has recently been reported to be of potential benefit for risk stratification and as a surrogate biomarker for individual susceptibility for anthracycline-induced cardiotoxicity (17). In a small study, the level of topoisomerase IIβ was significantly higher in the anthracycline-sensitive group. Close cardiac monitoring with early recognition and treatment of LV dysfunction using β-blockers and ACE inhibitors within the first 6 months of onset has been shown to be associated with stabilization and even recovery of cardiac function (18). These observations are the basis for the current recommendations of routine cardiac monitoring during anthracycline therapy. Different diagnostic tools have been useful in monitoring these patients including the use of cardiac biomarkers and/or cardiac imaging studies (19). There is no consensus regarding the best approach and optimal timing for testing. At the University of Texas MD Anderson Cancer Center (MDACC), we rely on serial imaging with two-/three-dimensional echocardiograms with myocardial strain to monitor these patients (Figs. 54-1 and 54-6).
FIGURE 54-6
Algorithm for cardiac monitoring and management of patients receiving potentially cardiotoxic chemotherapy. 2D/3D, two-dimensional/three-dimensional; GLS, global longitudinal strain; HF, heart failure; LVEF, left ventricular ejection fraction; TKI, tyrosine kinase inhibitor. aEquivalent cardiotoxicity dose to 300 mg/m2 of doxorubicin = 150 mg/m2 of idarubicin = 900 mg/m2 of epirubicin.

Multiple primary preventive measures may be needed to lower the risks of anthracycline-induced cardiomyopathy. Continuous infusions instead of repetitive boluses are associated with a lower incidence of myocyte damage (20). Drug peak plasma level correlates with the degree of myocyte toxicity, whereas the area under the curve determines antitumor efficacy. Modified preparations of anthracyclines such as liposomal doxorubicin are associated with lower risk. Dexrazoxane is thought to lower risk through iron chelation and interference with the topoisomerase IIβ complex by preventing it from binding to anthracyclines. Finally, the use of β-blockers and ACE inhibitors or angiotensin receptor blockers has been associated with mixed results in small prospective studies (21,22). It is not clear if they exert a potential cell protection effect or if they only have a beneficial hemodynamic effect. Their role in primary prevention of chemotherapy-induced cardiotoxicity is uncertain.
This toxicity is associated with reversible disruption of myocyte contractile function. Trastuzumab is the prototype drug. Other targeted chemotherapy agents, including several small-molecule oral TKIs, are also suspected to exert type II cardiotoxicity. The mechanism involves disruption of signaling pathways responsible for tumor growth and also for cardiac cell repair (ErbB2 in the case of trastuzumab), resulting in myocyte dysfunction but not cell death (23). Up to a third of patients treated with trastuzumab develop evidence of LV systolic dysfunction or HF symptoms that are often reversible upon discontinuation of this agent (24). Re-initiation or continuation of trastuzumab after LV systolic function recovery is usually well tolerated. The US Food and Drug Administration (FDA) recommends monitoring LV function with imaging every 3 months during therapy. Not all patients are at the same risk, and several predisposing factors increase susceptibility for trastuzumab cardiotoxicity. These include concomitant use of anthracyclines, advanced age, and HTN or other underlying structural heart disease. There is an incremental increase in the incidence of LV systolic dysfunction or HF as the number of risk factors increases. For example, the risk of myocardial dysfunction increases from <1% in young women with no risk factors to 27% in elderly patients receiving concomitant anthracyclines (12). A clinical risk stratification model (National Surgical Adjuvant Breast and Bowel Project cardiac risk score) has been proposed to help risk stratify patients, but it needs to be validated (25).
Tyrosine kinase inhibitor–related LV dysfunction has been reported since early phase I and II studies. The mechanisms of toxicity are not fully understood, and several theories have been proposed, including apoptotic versus nonapoptotic cell death, dysregulation of cellular energy hemostasis through inhibition of 5-AMPK (26), mitochondrial toxicity, and HTN-mediated LV dysfunction (27). The incidence of TKI-related cardiomyopathy and HF varies widely with different drugs (mostly reported with sunitinib, sorafenib, imatinib, nilotinib, and ponatinib) and differs between studies by almost 10-fold. This is partially related to the fact that cancer trials typically do not have cardiovascular events as outcomes, and this likely leads to an underestimation of the incidence of such events. Also, the usual clinical HF symptoms of fatigue, dyspnea, and leg edema are nonspecific in patients with cancer, leading to limitations in clinical diagnostic accuracy. The incidence of symptomatic HF necessitating medical therapy varies between 1.5% and 15%, and the rate of reported drop in LVEF is between 7% and 28% (27,28,29,30,31). Time to onset of cardiomyopathy varies from a few weeks to several months after initiation of TKIs. Evidence of reversibility of significant LV dysfunction was seen in half of the patients after TKIs were discontinued.
When clinical HF or LV systolic dysfunction is confirmed, patients typically are taken off of the TKI, and HF drugs are initiated (ACE inhibitors, β-blockers, and diuretics). Depending on the patient risk factors and clinical presentation, workup for other possible etiologies of cardiomyopathy should be performed when appropriate (ie, ischemic heart disease, HTN). In the absence of strong prospective data, there is not enough information to support the routine use of cardiac imaging to monitor asymptomatic patients. It is common practice to obtain a baseline two-dimensional echocardiogram study before initiating these drugs, with a low clinical threshold to obtain repeated echocardiographic studies if dyspnea or fluid retention symptoms develop on TKIs.
This is usually observed in patients with underlying diastolic or systolic LV dysfunction including hypertensive heart disease, ischemic heart disease, or nonischemic cardiomyopathy. Decompensated HF symptoms are often triggered by large amounts of intravenous fluids given during certain chemotherapy infusions (eg, cisplatin). Patients typically respond well to withholding fluids and using diuretics. Standard HF therapy, including β-blockers and ACE inhibitors, is indicated if LV systolic dysfunction is present (32).
Infiltrative processes are another cause of cardiomyopathies commonly encountered. Examples include amyloidosis and secondary hemochromatosis. In patients with myelodysplastic syndrome, myocardial iron overload develops from frequent blood transfusions (ie, >100 units).
The association between malignancy and ischemic arterial diseases is well established. The clinical presentation and management of arterial ischemic events vary based on the arterial bed and the organ involved. The clinical spectrum includes stroke, myocardial infarction, and visceral and limb ischemia. In two large separate cohorts of patients with cancer, Khorana et al reported a 1.5% to 3.1% incidence of arterial ischemic events (33,34). The most common events were cardiac, and less than 0.5% of events involved limb ischemia. The rate of arterial ischemic events is higher in specific cancer populations such as those with myeloproliferative disorders or hematologic malignancies with secondary amyloidosis. To assess the significance and outcome of these arterial events, Khorana et al prospectively followed 4,466 patients receiving active chemotherapy. Thromboembolism was a leading cause of death (9.2%) (35), with a higher rate of death from arterial events compared to venous events.
In addition to the usual causes and the traditional risk factors typically associated with arterial ischemia in the general population, patients with underlying malignancy have added increased risks for arterial ischemic events related to the inherent thrombophilia associated with the cancer and its therapy (Table 54-3). From the clinical perspective, it is useful to divide these cancer-related etiologies into two broad categories; the first category includes mechanisms, and the second group includes cancer etiologies.
| Noncancer Patients | Cancer Patients | |
Atherosclerosis Atrial fibrillation Aortic arch plaques Mitral stenosis Valvular prosthesis Infective endocarditis Thrombophilia Antiphospholipid syndrome Antithrombin deficiency Protein C and S deficiency | Paraneoplastic syndrome Tumor invasion Tumor embolization Paradoxical embolization | Leukemia Amyloidosis Myeloproliferative disorders Chemotherapy Radiation therapy |
Multifactorial mechanisms have been implicated in the pathogenesis of hypercoagulability and thrombosis. As shown in Fig. 54-7, circulating and in situ cancer cells can enhance activity of tissue factor and other cancer procoagulant factors and can activate platelets. These mediators can then trigger coagulation in previously damaged vessels like the coronary arteries or peripheral arteries or even in previously healthy vessels (36). The end result is cancer-enhanced thrombosis, which manifests as (1) low-grade disseminated intravascular coagulation, (2) venous thrombophlebitis, (3) arterial thrombosis, (4) accelerated ischemic cardiac and peripheral vascular disease, and (5) nonbacterial thrombotic endocarditis. Another subgroup of patients with a hypercoagulable condition present with digital ischemia with no evidence of large-vessel involvement (Fig. 54-8). The mechanism is thought to be due to capillary deposition of antigen-mediated antibody complexes from tumor cells. This paraneoplastic syndrome is usually very difficult to treat; symptoms do not respond to usual vascular therapy until the cancer is fully controlled (37).
FIGURE 54-7
Suggested mechanism for the enhanced thrombosis observed in cancer patients. DIC, disseminated intravascular coagulation; IHD, ischemic heart disease; NBTE, nonbacterial thrombotic endocarditis; PVD, peripheral vascular disease.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


