Historically, acute leukemias that do not fit neatly into the broad categories of “acute myeloid leukemia” or “acute lymphoblastic leukemia” have caused considerable confusion and frustration for clinicians and pathologists. These misfit cases include bilineage and biphenotypic leukemias (collectively termed “mixed phenotype acute leukemias” in the 2008 World Health Organization Classification of Tumors of Hematopoietic and Lymphoid Tissues), as well as acute undifferentiated leukemias and other miscellaneous atypical cases. In this review, the author discusses the definitions, history, clinical behavior, and current treatment approaches to acute leukemias of Sambiguous lineage.
Using currently available diagnostic tools, skilled hematopathologists can assign most pediatric and adult acute leukemias to myeloid, B-lymphoid, or T-lymphoid lineages without much difficulty. However, a subset of more challenging cases (depending on the particular criteria used for lineage assignment, this subset may represent as many as 5% of all acute leukemias) cannot be easily classified, despite integration of data from flow cytometric, immunohistochemical, cytochemical, cytogenetic, and molecular genetic investigations.
This uncommon and heterogeneous group of acute leukemias has received a degree of attention disproportionate to its prevalence, and has been described using a variety of terms: bilineage , biphenotypic , hybrid , polyphenotypic , mixed lineage , mixed phenotype , ambiguous lineage , uncertain origin . The most recent (2008, fourth edition) World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid Tissues includes 7 categories of varying specificity within an “acute leukemias of ambiguous origin” diagnostic group, including bilineage and biphenotypic leukemias, which are classified together as “mixed phenotype acute leukemias” (MPAL) ( Table 1 ). The WHO ambiguous-origin leukemias also include an “other ambiguous lineage leukemias” category, which contains a provisional entity, “natural killer cell lymphoblastic leukemia/lymphoma,” as well as 2 categories defined by both immunophenotype and by one of a pair of specific molecular genetic findings, BCR-ABL fusion and MLL rearrangement. With the exception of BCR-ABL1 fusion, the oncogenic protein product of which can be inhibited using targeted tyrosine kinase inhibitors, the clinical relevance of the other WHO-defined ambiguous leukemia distinctions remains unclear.
| Category | Definition and Comments |
|---|---|
| Acute undifferentiated leukemia | An acute leukemia that does not express any marker that is considered specific for either lymphoid or myeloid lineage . a Nonhematopoietic tumors (eg, germ cell tumors) and acute leukemias of unusual lineage (eg, those derived from myeloid or plasmacytoid dendritic cell precursors, NK-cell precursors, basophils) must be excluded. Blasts may express HLA-DR, CD34, or CD38 and may be positive for TdT. |
| Mixed phenotype acute leukemia with t(9;22)(q34;q11.2); BCR-ABL1 | An acute leukemia that meets diagnostic criteria b for MPAL, in which the blasts also have the t(9;22) chromosome translocation or the BCR-ABL1 rearrangement . Patients known to have had chronic myeloid leukemia before developing blast phase should not be assigned to this category, even if they meet diagnostic criteria for MPAL. |
| Mixed phenotype acute leukemia with t(v;11q23); MLL rearranged | An acute leukemia that meets diagnostic criteria b for MPAL, in which the blasts also have a translocation involving the MLL gene at 11q23 . Cases of ALL that express myeloid antigens but do not meet criteria for MPAL should not be assigned to this category. |
| Mixed phenotype acute leukemia, B/myeloid, NOS | An acute leukemia that meets diagnostic criteria for assignment to both B and myeloid lineage , in which the blasts lack genetic abnormalities involving BCR-ABL1 or MLL |
| Mixed phenotype acute leukemia, T/myeloid, NOS | An acute leukemia that meets diagnostic criteria for assignment to both T and myeloid lineage , in which the blasts lack genetic abnormalities involving BCR-ABL1 or MLL |
| Mixed phenotype acute leukemia, NOS – rare types | An acute leukemia with a mixed phenotype that is not listed above. Examples would include a lymphoid leukemia with both B and T lineage commitment, or a leukemia with evidence of trilineage (ie, B/T/myeloid) markers. Erythroleukemias and megakaryocytic leukemias are clearly of myeloid origin, but do not typically express MPO, so B/ or T/erythroleukemia, or B/ or T/megakaryocytic leukemia, if they exist (they had not yet been described at the time of the 2008 WHO classification, but a case report of B/erythroleukemia appeared shortly thereafter ), would fit in this category. |
| Other ambiguous lineage leukemias | An acute leukemia that expresses an unusual combination of markers that does not allow classification as either AUL, MPAL, or a single lineage leukemia ( eg, a form of AML or ALL) using WHO diagnostic criteria c for lineage assignment. This category includes a rare provisional entity: (Precursor) natural killer cell lymphoblastic leukemia/lymphoma, which expresses CD56 along with immature T-associated markers, and lacks B-cell and myeloid markers as well as T-cell clonal gene rearrangement. CD94 1A and CD161 might be expressed, but currently are rarely tested in clinical practice. |
a By definition, acute undifferentiated leukemias lack cytoplasmic CD3 (T lineage defining), lack MPO (myeloid lineage defining), do not express B-cell–specific markers (eg, cytoplasmic CD22, cytoplasmic CD79a, or strong expression of CD19), and lack specific features of other lineages, such as plasmacytoid dendritic cells, erythroblasts, or megakaryocytes. For further discussion of lineage-specific markers, see Tables 2 and 3 and relevant text.
b Diagnostic criteria for “mixed phenotype acute leukemia” require assignment of blast cells to 2 or 3 lineages, using lineage-defining criteria in Table 3 . Mixed phenotype may result from either 2 distinct blast cell populations or 2 or more lineage-associated markers on the same blast cells.
c These cases must be distinguished from blastic plasmacytoid dendritic cell neoplasm (BPDCN), which was not clearly part of the 2001 WHO classification but is included as a distinct entity in the 2008 WHO classification. BPDCN tumor cells express CD4, CD56, CD43, CD45RA, and the plasmacytoid dendritic cell neoplasm markers CD123, CD303 (BDCA2), TCL1, CLA, and MxA; 50% of cases express CD68, whereas CD7, TdT, and CD33 expression are common. Names used in the past for this neoplastic entity (BPDCN) include the following: blastic NK cell lymphoma, agranular CD4+ NK cell leukemia, blastic NK leukemia/lymphoma, or agranular CD4+ CD56+ hematodermic neoplasm/tumor. These cases also must be distinguished from myeloid/NK cell acute leukemia, which is a form of AML with minimal differentiation, possibly of precursor NK cell origin (but early NK cells express no specific markers).
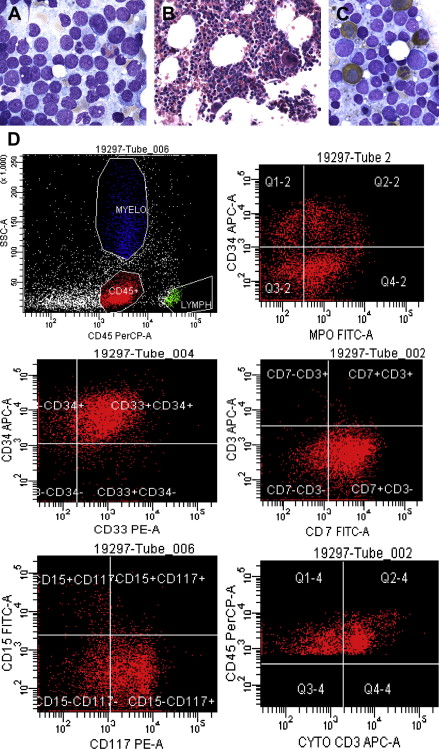
The general perception among clinicians is that whether they appear in adults or children, leukemias of ambiguous origin are as difficult to treat successfully as they are to classify definitively ( Fig. 1 ). Indeed, in most published series, outcomes are poorer for leukemias of ambiguous origin than for more typical cases of acute myeloid leukemia (AML) or acute lymphoblastic leukemia (ALL), and allogeneic hematopoietic stem cell transplantation is commonly offered to transplant-eligible patients, albeit more as an act of desperation in the face of extremely high relapse risk than as a data-driven care choice. Improved treatment approaches will almost certainly require moving beyond immunophenotyping and cytochemistry, and toward better understanding of the molecular pathobiology of these uncommon disorders. The evolution of diagnostic criteria, current understanding, and potential approaches to therapy for leukemias of ambiguous lineage are reviewed in this article.
Historical developments and diagnostic criteria
Developments Before 1991
Because lineage infidelity in leukemic blasts could not be described until lineage-associated markers became widely available, recognition of the more peculiar varieties of leukemia is, in the history of hematological oncology, a relatively recent development. This development is closely tied to the advent of routine cytochemical evaluation of marrow specimens in the 1970s, introduction of monoclonal antibodies in the early 1980s, and increasing use of multiparameter flow cytometry in leukemia phenotyping during the late 1980s and early 1990s.
Before 1980, clinicians and morphologists assigned leukemia diagnoses on the basis of clinical presentation and cell morphologic features, such as the cytoplasmic appearance and presence or absence of Auer rods, supplemented by basic cytochemical stains, such as Sudan Black B, chloroacetate esterase, and the periodic acid-Schiff reaction. The lineage and corresponding cytologic appearance and immunophenotype of blast cells in acute leukemia was believed to cleanly reflect malignant transformation and maturation arrest at a particular stage of hematopoietic differentiation, with acute lymphoid malignancies originating in lymphoid progenitors and acute myeloid malignancies originating in early myeloid series cells.
Several observations in the 1980s challenged this assumption, however. Expression of the terminal deoxynucleotidyl transferase (TdT) enzyme, thought to be an exclusive marker of lymphoid lineage for several years following its discovery by McCaffrey and colleagues in blasts from children with ALL in 1973, was found by 1981 to occur in 10% to 20% of cases of AML. (In fact, if more sensitive techniques are used, TdT activity is detectable in blasts in as many as 55% of AML cases, albeit usually at a much lower level of expression than is typically seen in TdT-positive ALL. ) Similarly, in the 1980s morphologists also learned that myeloperoxidase (MPO) expression, considered to be a myeloid lineage-defining marker after its initial description in the 1940s, is detectable by either cytochemistry or immunohistochemistry in more than 20% of B-ALL cases.
Additionally, in the late 1970s and early 1980s, a series of case reports and small series appeared in which investigators described detecting either 2 distinct populations of blasts of differing putative origin simultaneously in the same patient (“bilineage” leukemias), or a homogeneous population of blasts expressing markers of more than one lineage (“biphenotypic” leukemias). The relationship between bilineage and biphenotypic leukemias remained unclear, and in some instances leukemic blasts that expressed markers most suggestive of one cell differentiation program at the time of initial diagnosis would subsequently exhibit a different immunophenotype consistent with another lineage at relapse (eg, B-cell ALL at diagnosis, but AML without lymphoid lineage markers at relapse following ALL-directed chemotherapy).
In 1985, Mirro and colleagues assessed 123 consecutive pediatric leukemia cases diagnosed at St Jude Children’s Research Hospital in Memphis, Tennessee, or in Calgary, Alberta, Canada, using a panel of myeloid and lymphoid-associated antibodies, and found that ALL blasts coexpressed at least one myeloid marker in 19% of cases, whereas the leukemic blasts in 25% of AML cases expressed at least one lymphoid-associated marker. Although this landmark study highlighted the frequency of lineage infidelity in leukemia, the antibodies used to assign lineage in the study by Mirro and colleagues (ie, CD2 [T-11 antibody], CD3e [T-3], CD5 [T-101], and CD10 [J5 CALLA] for lymphoid; CD11b [Mo1 antibody], CD13 [MCS.2 and SJ-D1], CD15 [My-1], and CD36 [5F1] for myeloid) would, with the exception of CD3e, no longer be considered lineage-defining markers.
Investigators compiling adult leukemia series soon noted that a large proportion of patients with AML or ALL express at least one lineage infidelity marker, but that the presence of only a single such antigen does not alter clinical behavior or response to treatment. Further complicating matters, some markers initially thought to be tightly lineage-associated later proved to be so commonly expressed in malignancies of other lineages as to no longer even be considered aberrant. One such example is the CD15 carbohydrate adhesion molecule, which is expressed on normal neutrophils and on blast cells in most AML cases so was once considered myeloid-associated, but is also very commonly detected in B-cell ALL lacking CD10 expression and in other non-myeloid neoplasms (eg, Hodgkin lymphoma).
Development of European Group for the Immunologic Characterization of Leukemias Criteria
A growing number of case reports and small series in the 1980s highlighted increasing diagnostic confusion related to mixed lineage acute leukemias, and the need for more formal definitions of these somewhat puzzling disorders became increasingly clear. Catovsky and a group of European colleagues proposed the first scoring system for defining biphenotypic acute leukemia (BAL) in 1991, which weighed various cytochemical/immunohistochemical and immunophenotypic markers according to their perceived lineage specificity.
In 1995, the European Group for the Immunologic Characterization of Leukemias (EGIL) refined this 1991 system ( Table 2 ) to make it somewhat more stringent, requiring a lineage-associated marker score of more than 2 points in each of 2 lineages to define BAL, as well as demoting several markers that Catovsky and colleagues had proposed as supporting lineage assignment (ie, immunoglobulin heavy chain gene rearrangement for B lineage; CD11b/c and CD12 for myeloid lineage) while adding or refining several others (ie, CD20 and CD79a for B lineage; TdT, CD1a, CD7, CD8, and CD10 for T lineage; CD13, CD64, and CD65 for myeloid lineage). The EGIL system was modified in 1998 to add CD117 as a putative myeloid-specific marker, but soon a few ALL cases were found that expressed CD117.
| Score | B Lineage | T Lineage | Myeloid Lineage |
|---|---|---|---|
| 2 points |
|
|
|
| 1 point |
|
|
|
| 0.5 point |
|
|
|
Clinicians and investigators quickly raised concerns about the EGIL criteria, such as the lack of specificity of certain EGIL-proposed markers, especially CD79a expression for defining B lineage and MPO positivity for defining myeloid lineage. In addition, the EGIL system used varying and arbitrary thresholds of cellular antigen expression required to qualify as “positive” (eg, 20% for most markers, but 10% for MPO, CD3, TdT, and CD79a), did not distinguish between biphenotypic and bilineage leukemia, did not assess intensity of antigen expression, and also failed to incorporate cytogenetic data. The latter proved to be a particularly troublesome limitation, because leukemia subtypes that were already well defined by routine G-banded karyotyping by the time the EGIL system was introduced (and, later, were further clarified using molecular genetic techniques) often express a degree of lineage infidelity, yet have other uniform features or may respond similarly to therapy regardless of immunophenotype. For example, t(8;21) AML often expresses a subset of B-cell antigens, whereas acute promyelocytic leukemia (APL), especially the hypogranular variant, frequently expresses T-cell markers, particularly CD2, and T-cell marker expression in APL has no effect on response to all-trans retinoic acid therapy. In addition, CD79a can be found in both T-ALL and myeloid leukemia, so lacks the B-cell specificity accorded to it by the EGIL system.
World Health Organization Criteria: Third and Fourth Editions
In the third edition of the WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues proposed in 1999 and published in 2001, the WHO working group included a 2-page description of a classification of “acute leukemias of ambiguous lineage,” composed of 3 categories: bilineage, biphenotypic, and undifferentiated acute leukemias. Although the third edition WHO classification referenced the 1995 EGIL system (with the 1998 CD117 addition), the WHO third edition text unfortunately included a typographical error in its Table 4.03, stating that only 2 points, rather than more than 2 points, were necessary to assign a leukemia case to a given lineage. In one series, these slightly less stringent criteria tripled the number of cases defined as biphenotypic compared with EGIL criteria, and the reassignment was of dubious clinical value.
Perhaps because of this debacle, the 2008 fourth edition of the WHO classification did away with lineage scoring entirely in favor of descriptive but relatively specific requirements for assigning more than one lineage to a single blast population ( Table 3 ). Although the WHO fourth edition classification retained the category of acute leukemias of ambiguous lineage, the updated system also incorporated 5 major changes with respect to the third edition classification.
| Lineage | Criterion #1 | Criterion #2 |
|---|---|---|
| B lineage | Strong CD19, plus at least 1 of CD79a (cytoplasmic or membrane), cytoplasmic CD22, or surface CD10 also strongly expressed | Weak CD19, plus at least 2 of CD79a, cytoplasmic CD22, CD10 |
| T lineage | Cytoplasmic expression of CD3 (detected by flow cytometry with antibodies against the epsilon chain of CD3 a ) | Surface CD3 (rare) |
| Myeloid | Myeloperoxidase positivity in >3% of blasts (detected by flow cytometry, immunohistochemistry, or cytochemistry) | At least 2 of the following monocytic differentiation markers: CD11c, CD14, CD64, lysozyme, nonspecific (butyrate) esterase |
a Immunohistochemistry using polyclonal anti-CD3 antibodies is not adequate, as this assay may detect CD3 zeta chain, which is not T-cell specific.
First, because of frequent ambiguity about whether mixed lineage markers are truly coexpressed by the same blasts or by 2 distinct populations of leukemic cells, WHO did away with the terms “bilineal” and “biphenotypic” in favor of the collective term “mixed phenotype acute leukemia” (MPAL). Second, the criteria that define the myeloid, T-lymphoid, and B-lymphoid components of MPAL were altered and simplified (see Table 3 ), although the same criticisms persisted about lineage specificity of proposed markers. Third, cases that are clearly acute leukemia with recurrent genetic abnormalities were excluded from MPAL, even if they express a mixed lineage phenotype that would otherwise qualify. Such entities include cases classified as AML with recurrent genetic abnormalities (eg, t[15;17], t[8;21], and inv[16]), but the WHO committee did not feel comfortable extending this “trumping” of immunophenotype by genetic and molecular markers to provisional entities, such as AML with mutated NPM1 or CEBPA. Similarly excluded regardless of lineage expression are cases known to represent chronic myeloid leukemia (CML) in blast phase, therapy-related AML, or AML arising from a myelodysplastic syndrome.
The fourth change between third and fourth editions of the WHO classification was inclusion of 2 molecularly defined diagnostic categories in the acute leukemia of ambiguous lineage group: BCR-ABL1 –positive and MLL -rearranged acute leukemias. In many series of EGIL-defined BAL, t(9;22) and t(11q23) translocations have been overrepresented, probably because loss of ABL tyrosine kinase activity or MLL fusions with associated HOX dysregulation severely disrupts normal differentiation programs. Indeed, the MLL gene took its name from its association with “mixed lineage leukemia” or “myeloid-lymphoid leukemia.”
Unfortunately, although well intentioned, these molecularly defined acute leukemia of ambiguous origin categories quickly caused confusion. MLL-rearranged acute leukemias without a mixed lineage phenotype are elsewhere classified by WHO both as “AML with t(9;11)(p22;q23); MLLT3-MLL” and variants, and as “B lymphoblastic leukemia/lymphoma with t(v;11q23); MLL rearranged.” These entities may be seen with a much greater frequency to MPAL with MLL rearrangement, and the prognosis and treatment are determined by the genotype rather than the immunophenotype.
In addition, patients who previously had unrecognized chronic myeloid leukemia (CML) and first present to medical attention at a late stage (ie, in blast crisis), are not easy to distinguish from those with BCR-ABL1 –positive de novo ALL or AML, as p190 and p210 transcripts can be detected in either setting. Although the WHO classification states, “this diagnosis (ie, MPAL with t[9;22][q34;q11.2; BCR-ABL1]) should not be made in patients known to have had CML,” the stage at which the patient presents for medical attention is a poor criterion on which to base a disease classification, as delay in diagnosis does not reflect a genuine biologic difference and may be because of socioeconomic or other personal factors.
A fifth change between the 2001 and 2008 classifications is that in the 2008 edition, the WHO committee also included an “other ambiguous lineage leukemias” category for truly bizarre cells. For instance, leukemia cases that express T-cell lineage markers but not cytoplasmic CD3, or cases that express several myeloid markers but not MPO, do not fit into any of the other MPAL categories.
These cases are considered distinct from cases with no lineage-specific markers at all, which WHO designates as acute undifferentiated leukemia (AUL) and should be diagnosed only after a cautious, thorough evaluation. AUL cases should express CD34, HLA-DR, or CD38—something to exclude the possibility of a nonhematopoietic neoplasm—but, by definition, lack specific myeloid or lymphoid antigens. Some AUL cases have a t(9;9) translocation that results in fusion of SET, a nuclear phosphoprotein that inhibits activity of protein phosphatase 2A (PP2A), to the oncoprotein CAN/NUP214 involved in the more common t(6;9) in AML.
Finally, the difficult-to-define entity of natural killer cell lymphoblastic leukemia/lymphoma was included as a provisional category within the miscellaneous “other ambiguous lineage leukemias” category in the fourth WHO classification, whereas those cases more clearly designated as originating from plasmacytoid dendritic cells (also CD56 positive, like natural killer cells) were reclassified as a new entity, blastic plasmacytoid dendritic cell neoplasms (BPDCNs). By definition, BPDCNs coexpress CD4 and CD56, and usually also express CD123 or CD303 (BDCA2) (see Table 1 footnotes).
Limitations of Current Criteria
In addition to the concerns raised about lineage specificity of particular markers and variable intensity of marker expression, clinicians have expressed concerns that to some extent trying to pigeonhole leukemias of ambiguous origin is an exercise in futility. It is not clear whether, for example, T/myeloid leukemia and B/myeloid leukemia are genuinely distinct conditions requiring a different therapeutic approach, and whether the WHO 2008 criteria are really defining “real” entities in the sense of clinically actionable unique disorders.
There are also technical concerns. Currently, pathologists primarily perform immunophenotyping of new leukemia cases by flow cytometry, and flow is to some extent operator-dependent, with results varying depending on such parameters as gating and reagent quality. In addition, most clinical laboratories, even those at major academic centers, normally do not use all the flow markers in the EGIL panel or even all of the major WHO markers, so biphenotypic/bilineage expression may be missed. A conference convened in 2006 in Bethesda, Maryland, defined a consensus set of reagents suitable for general use in the diagnosis and monitoring of hematopoietic neoplasms, but did not recommend the full panel necessary for defining biphenotypic leukemia according to EGIL criteria. For example, many laboratories do not routinely perform cytoplasmic CD79a as part of a standard new leukemia diagnostic flow panel, in part because as stated, this “B-lineage–specific” marker also is regularly found in cases of AML (especially t[8;21] or APL ) and T-ALL.
Related posts:
 Induction and Postremission Strategies in Acute Myeloid Leukemia: State of the Art and Future Directions
Prognostic Factors in Adult Acute Leukemia
Curing All Patients with Acute Promyelocytic Leukemia: Are We There Yet?
Acute Leukemias
Clinical Implications of Novel Mutations in Epigenetic Modifiers in AML
Novel Transplant Strategies in Adults with Acute Leukemia
Induction and Postremission Strategies in Acute Myeloid Leukemia: State of the Art and Future Directions
Prognostic Factors in Adult Acute Leukemia
Curing All Patients with Acute Promyelocytic Leukemia: Are We There Yet?
Acute Leukemias
Clinical Implications of Novel Mutations in Epigenetic Modifiers in AML
Novel Transplant Strategies in Adults with Acute Leukemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


