Obesity
Eleftheria Maratos-Flier
Jeffrey S. Flier
Adipose tissue has three primary functions. Two are well recognized and defined: to serve as the site for storage of energy-rich fatty acids in the form of triglyceride and to effect the controlled release of the constituent fatty acids and glycerol in response to neural, endocrine, and local signals for metabolism at distant sites. A third and increasingly important role of adipose tissue is its function as an endocrine organ, releasing a variety of factors that regulate metabolism. Obesity is defined as a state of excessive adipose tissue mass and is best viewed as a syndrome or group of diseases rather than as a single disease entity. The importance of this state derives from its high prevalence in our society and its association with serious morbidity, not the least of which is a marked increase in the prevalence of type 2 diabetes. Specific syndromes of obesity, both in animal models and in humans, are associated with identified neural, endocrine, or genetic causes. However, the pathogenesis of obesity in the vast majority of humans is unknown and remains an unexplained chronic excess in caloric intake relative to energy needs. An understanding of obesity and its consequences requires the investigation of the many factors that control energy intake and energy expenditure, the two interrelated components of the energy-balance equation. As with our understanding of pathogenesis, our understanding of the molecular connection between obesity and its most important complications is limited, and our approach to therapy, as defined by clinical success rate, is extremely poor. In this chapter, we will review the current understanding of the pathogenesis, complications, and treatment of obesity.
DEFINITION AND INDICES OF OBESITY
The distribution of body weight in the population is a continuous function without a clear separation between lean and obese. The most medically relevant criterion relates to the identification of a weight that confers morbidity. The selection of a specific threshold is somewhat arbitrary, and a number of different criteria have been used. Initially, the approach was through the use of life insurance data (Metropolitan Life) that assess mortality as a function of body weight per height, adjusted for frame size, with obesity defined on purely statistical grounds as a weight that is 20% or more above the average weight per height (1). Over the past decade, calculation of body mass index (BMI) has evolved as a more standard measurement used to correlate weight with morbidity and mortality. BMI is calculated by determining weight in kilograms and dividing by the height in meters squared. This measurement has been used to define four classes of body weight. A BMI of less than 18.5 is considered underweight and carries a modestly increased risk of morbidity
and mortality. A BMI between 18.5 and 24.9 is considered normal. A BMI of more than 25.0 but less than 29.9 is considered overweight or preobese and, statistically, carries a slightly increased risk of comorbidities such as diabetes and cardiovascular disease compared with the risk in normal-weight individuals. A BMI of more than 30 is considered in the obese category, which is further subdivided into class I (BMI, 30 to 39.9), class II (BMI, 40 to 49.9), and class III (BMI, >50). These categories of obesity carry respective risks of comorbidities that are moderate, severe, and very severe, respectively (2). A second approach to defining the obese state involves quantitation of adipose tissue, either directly or indirectly. Values are obtained for a reference group viewed as normal, and obesity is defined as levels of adiposity exceeding that seen in the reference group.
and mortality. A BMI between 18.5 and 24.9 is considered normal. A BMI of more than 25.0 but less than 29.9 is considered overweight or preobese and, statistically, carries a slightly increased risk of comorbidities such as diabetes and cardiovascular disease compared with the risk in normal-weight individuals. A BMI of more than 30 is considered in the obese category, which is further subdivided into class I (BMI, 30 to 39.9), class II (BMI, 40 to 49.9), and class III (BMI, >50). These categories of obesity carry respective risks of comorbidities that are moderate, severe, and very severe, respectively (2). A second approach to defining the obese state involves quantitation of adipose tissue, either directly or indirectly. Values are obtained for a reference group viewed as normal, and obesity is defined as levels of adiposity exceeding that seen in the reference group.
The definition of obesity can be refined on the basis of the realization that the accumulation of adipose tissue in different depots has distinct consequences. Thus, many of the most important complications of obesity, including insulin resistance, diabetes, hypertension, and hyperlipidemia, are linked to the amount of intraabdominal fat, rather than to lower-body fat (i.e., buttocks and leg) or subcutaneous abdominal fat (3,4). Abdominal fat, typically evident on physical examination, can be estimated by determining the waist-to-hip circumference ratio (with a ratio >0.72 considered abnormal), or more accurately quantified by dual-energy x-ray absorptiometry (DEXA) scanning or computed tomography.
PREVALENCE OF OBESITY
It is obvious from casual inspection of the population that obesity is prevalent in the United States, although the precise prevalence figures vary to some degree, depending on the nature of the population surveyed. In the United States, the prevalence of overweight or preobesity, i.e., a BMI of 25 to 29.9, has remained fairly constant at 40% for men and 24% for women over roughly a 30-year period (1960 to 1994). However, in the same period, the prevalence of a BMI higher than 30 has risen significantly, especially over the past decade. In 2001 more than 20% of all adults had a BMI greater than 30, compared with 12% of all adults in 1991. Obesity among adults aged 18 to 29 doubled from 7% in 1991 to 14% in 2001. Among people aged 50 to 59, more than 25% have a BMI greater than 30. When analyzed by race and ethnicity (Fig. 31.1A), a BMI greater than 30 was most prevalent among black, non-Hispanic people. Interestingly, rates of obesity correlate inversely with educational level and are almost twice as high in adults who have not completed high school as in adults who have finished college or graduate school (Fig. 31.1B). This indicates that environmental and cultural factors can act as inhibitors to weight gain. At present 60% of the male population and 50% of the female population have a BMI greater than 25, which is associated with increased risk of morbidity and mortality.
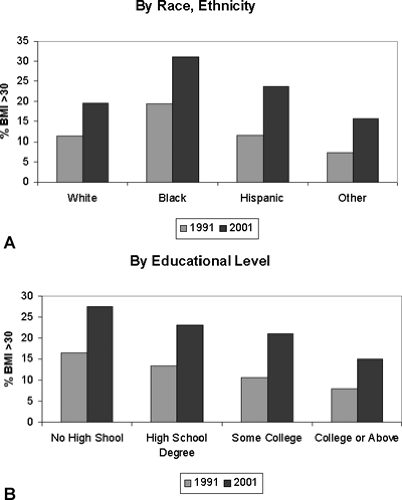 Figure 31.1. Prevalence rates of normal weight, overweight, and obesity in the adult U.S. population in 1991 and 2001. A: Rate of obesity by race, ethnicity. B: Rate of obesity by educational level. (Adapted from data from the Centers for Disease Control and Prevention.) |
As noted above, an important predictor of the morbidity and mortality associated with obesity is the quantity of visceral fat. A rough index of the relative amounts of visceral and abdominal fat is the waist-to-hip ratio. The alternative patterns of body-fat distribution have been described as pear shaped (low waist-to-hip ratio) and apple shaped (higher waist-to-hip ratio). When the waist-to-hip ratio is less than 0.8, the relative risk of morbidities associated with obesity is lower than when the waist-to-hip ratio is greater than 1.0. Hence, the metabolic syndrome, which is a clustering of obesity and other cardiovascular risk factors, is more likely to be associated with visceral obesity (5).
PATHOLOGIC CONSEQUENCES OF OBESITY
Obesity results in morbidity and mortality largely because of its association with other diseases, including diabetes, cardiovascular disease, hypertension, sleep apnea, endometrial cancer, colon cancer, and gallbladder disease. Overall, in the United States, the excess mortality of obesity accounts for 300,000 deaths per year. It was estimated that the total spent for both weight reduction and treatment of the consequences of obesity was $100 billion in the United States in 2001. This represents 5.5% to 7.0% of all medical expenses (3).
Diabetes
The increased risk for type 2 diabetes in individuals with obesity is considerable. In persons aged 20 to 44, obesity is associated with a fourfold increase in the relative risk of diabetes (4). In a study of a cohort of more than 50,000 U.S. male health professionals, the risk of diabetes correlated strongly with BMI. In men with a BMI of 35 or higher, the multivariate relative risk of diabetes was 42.1 compared with the risk in men with a BMI of less than 23. BMI appears to be the dominant risk factor for type 2 diabetes. Even men with average relative weight had a significant increase in risk when compared with men in lower weight groups. A similar increased risk exists for women. Among 43,581 women enrolled in the Nurses’ Health Study, the relative risk for type 2 diabetes at the 90th percentile of BMI was 11.2 (6). Weight was the single most important predictor of diabetes. After adjustment for BMI, lack of exercise and a poor diet (i.e., foods with a high glycemic index and high in trans fat) were also associated with increased risk of diabetes (7). Another study examined new diagnoses of diabetes in a population between 18 and 44 years of age (8) and found an inverse
correlation with age and BMI. Adults developing diabetes before age 44 had an average BMI of 39, whereas adults developing diabetes at 45 or older had an average BMI of 33. Among all adults, the odds ratio for developing diabetes is 6.38 for those with a BMI greater than 40 (9). The results of these and other studies lend support to the concept that the vast majority of cases of type 2 diabetes could be prevented by the adoption of therapies and lifestyle characteristics that decrease obesity.
correlation with age and BMI. Adults developing diabetes before age 44 had an average BMI of 39, whereas adults developing diabetes at 45 or older had an average BMI of 33. Among all adults, the odds ratio for developing diabetes is 6.38 for those with a BMI greater than 40 (9). The results of these and other studies lend support to the concept that the vast majority of cases of type 2 diabetes could be prevented by the adoption of therapies and lifestyle characteristics that decrease obesity.
Although the precise mechanism by which obesity contributes to insulin resistance and type 2 diabetes has not yet been defined, it is likely related to the production of various factors derived from the adipocyte that act on fat, liver, or muscle to impair insulin action. Obesity is itself associated with hyperinsulinemia, and insulin may induce insulin resistance through downregulation of the insulin receptor. Potential candidate substances produced by fat that may cause insulin resistance include tumor necrosis factor and other cytokines, such as interleukin-6, and resistin and adiponectin. Increased levels of free fatty acids are also capable of inhibiting insulin action. It is intriguing that a recent report found that treatment with high-dose salicylate markedly improved insulin resistance, suggesting that obesity may induce an inflammatory state that contributes to insulin resistance (10).
Cardiovascular Disease
Obesity is an independent risk factor for cardiovascular disease (11), including coronary artery disease and congestive heart failure, in both men and women. Waist-to-hip ratio is the best predictor, and it is noteworthy that increased waist-to-hip ratio has an effect in women even at the relatively low BMI of 25. Visceral obesity is associated with increased occurrence of hypertension and an atherogenic lipid profile (12,13), both of which contribute to the development of cardiovascular disease. In addition, in the obese state, there is a need for perfusion of a greater mass of tissue, resulting in an increase in cardiac work. Blood volume, stroke volume, and cardiac output are all increased and result in increased ventricular mass, which is reversible with weight loss (14,15).
Pulmonary Disease
Abnormalities in pulmonary function may be seen in obese patients (16(17(18). These range from quantitative abnormalities in pulmonary function tests that have no established clinical significance to major dysfunction replete with symptoms and morbid consequences. The increased metabolic rate in obese subjects increases O2 consumption and CO2 production, and these changes result in increased minute ventilation. In subjects with marked obesity, the compliance of the chest wall is reduced, the work of breathing is increased, and the respiratory reserve volume and vital capacity are reduced; a resultant mismatch between ventilation and perfusion may result in hypoxemia. Severe obesity may cause hypoventilation, defined by the development of CO2 retention. The full designation of the obesity-hypoventilation, or pickwickian, syndrome includes somnolence, lethargy, and respiratory acidosis and typically also includes sleep apnea. Such patients may have reduced ventilatory drive to hypoxia and hypercapnia, as well as obstructive or mechanical causes of hypoventilation, and sleep studies may be necessary to distinguish among these.
Gallstones
Obesity is associated with enhanced biliary secretion of cholesterol. This results in supersaturation of bile and a higher incidence of gallstones—particularly cholesterol gallstones (19). Fasting, as opposed to more limited caloric restriction, increases the saturation of bile by reducing the phospholipid component, and cholecystitis induced by fasting is a well-recognized problem in obese individuals.
Cancer
Excess weight has been associated with increased rates of cancer. A recent study examining data for more than a million patients enrolled in the Cancer Prevention Study demonstrates convincingly that obese individuals are at increased risk for a number of cancers (20). The most dramatic increase in risk is seen for liver cancer. The relative risk of liver cancer was almost 2-fold higher in men with a BMI of 30.0 to 34.9 than in normal-weight individuals, and it was 4.5-fold higher in men with a BMI greater than 35. In men with a BMI higher than 35, the risk of stomach cancer was increased 1.94-fold, that of kidney cancer was increased 1.7-fold, and that of esophageal cancer was increased 1.6-fold over the risk in normal-weight individuals. The effect of obesity on cancers of the gastrointestinal tract was not as great in women, but the increase in relative risk in women was the same as that in men for kidney cancer. In women with a BMI greater than 35, the relative risk of cancer of the uterus was 2.8, of cancer of the cervix was 3.8, and of breast cancer was 1.7.
ENDOCRINE CONSEQUENCES OF OBESITY
Many alterations in endocrine function are seen in patients with established obesity. These changes can be induced by overeating, and normal function resumes after weight loss. Therefore, these changes are viewed as being secondary to the obese state. A possible causal link has been sought between some of these abnormalities and the pathogenesis of obesity, and thus they have undergone considerable scrutiny.
Endocrine Pancreas
As discussed earlier, hyperinsulinemia is a pervasive concomitant of obesity. Hyperinsulinemia results from an increased rate of insulin secretion (21), although patients with intraabdominal obesity may have decreased hepatic clearance of insulin (22). Hyperinsulinemia follows weight gain and reverses with weight loss and is most likely a consequence of insulin resistance that accompanies the obese state. Given the fact that, in the animal model of ventromedial hypothalamic lesions, hyperinsulinism driven by the vagus nerve may precede obesity (23), the possibility that a defect in central control of insulin secretion exists in a subset of persons with obesity should be considered. Studies of glucagon, somatostatin, pancreatic polypeptide, and amylin secretion in obesity have not been particularly revealing. However, more recent studies suggest that neuropeptides such as neuropeptide Y and melanocyte-stimulating hormone may have direct effects on the islet (24,25).
Thyroid
Given the known effect of thyroid hormone on basal metabolic rate, it is reasonable to speculate that defects in this axis might be a factor in obesity. In general, studies of obese individuals have revealed normal levels of thyroxine (T4) and thyroid-stimulating hormone (TSH) but increased levels of triiodothyronine (T3) in a minority of subjects. The increased T3 levels are probably secondary to increased carbohydrate intake, and they
decrease, as do values in nonobese subjects, in response to caloric restriction (26).
decrease, as do values in nonobese subjects, in response to caloric restriction (26).
Gonadal Function
Marked obesity in men is associated with changes in both testosterone and estrogen metabolism, although these are usually without clinical consequences. Rates of estrogen production, primarily from androgen precursors, are increased, as are levels of estradiol (27). Decreased total testosterone levels are commonly observed and appear to be secondary to diminished levels of sex hormone-binding globulin (SHBG), with a preservation of normal levels of free testosterone. Levels of free testosterone may, however, be reduced in men with massive obesity (28). These changes may result in gynecomastia.
In women, marked obesity is associated with increased androgen production, increased peripheral conversion of androgen to estrogen, an increased rate of estrogen production, and decreased levels of SHBG. This constellation may be a major cause of the amenorrhea not infrequently seen in morbidly obese women. Upper-body obesity is associated with increased testosterone production, decreased SHBG, and increased levels of free testosterone in comparison to levels in obese women with lower-body or gynoid obesity (29(30(31). The fact that upper-body obesity also is associated with hyperinsulinemia has led to the hypothesis that insulin may be a factor that contributes to hyperandrogenism through actions on the ovary, as seems to be the case in syndromes of extreme insulin resistance. The increased peripheral production of estrogen from androstenedione, which occurs to a greater degree in women with lower-body obesity, may contribute to the increased incidence of uterine cancer in obese postmenopausal women (32).
Adrenal Function
The relationship between obesity and altered adrenal function can be addressed from a number of perspectives. The first relates to the clinical issue of whether a given patient with obesity, particularly one with hypertension and glucose intolerance, has Cushing syndrome. In 90% of obese individuals, the overnight cortisol response to 1 mg of dexamethasone given at midnight is normal, a finding sufficient to rule out Cushing syndrome. The 10% of individuals who fail to suppress cortisol production adequately on this test will suppress cortisol production normally in the formal 2-day low-dose dexamethasone test (33).
Obesity, however, is commonly associated with abnormalities of the cortisol axis, with increases in the rates of cortisol production and levels of urinary 17-hydroxysteroids frequently observed. Despite these findings, serum cortisol levels—including their diurnal variation—appear to be normal, and no clear defects in adrenocorticotropic hormone (ACTH) secretion have been observed (34). Thus, the precise basis for the increased cortisol production is unclear. One reason for interest in this area is the finding that cortisol is overproduced in a number of animal models of obesity, such as the ob/ob mouse and the fa/fa rat, and that removal of the adrenal gland markedly ameliorates many of the phenotypic and biochemical findings in these animals (35,36). The role, if any, of increased cortisol production in human obesity has not been established.
Pituitary Function
Obesity is clearly associated with defects in growth hormone secretion (37,38). Levels of growth hormone in response to many stimuli, including insulin-induced hypoglycemia, arginine, levodopa, exercise, sleep, and the physiologic regulator growth hormone-releasing hormone, are reduced in obese individuals. Treatment with cholinergic antagonists may reverse this defect (39). Because administration of growth hormone will reduce the percentage of body fat (40), these observations raise the obvious question of whether functional growth hormone deficiency is present in obesity. On the basis of levels of insulin-like growth factor-1, this would appear not to be the case. Many investigations of pituitary adrenal function in obesity have been carried out, in part because of the persistent interest in whether subtle hypothalamic dysfunction might be present in this disorder. A variety of findings with no obvious clinical relevance have been made (37).
ETIOLOGY OF OBESITY
Maintenance of a normal body weight requires a match of food intake to energy expenditure. Chronic positive energy balance leads to storage of calories in fat, mostly as fat but also as increased lean body mass (41), whereas negative balance leads to utilization of stores, including energy stored as glycogen, fat, and lean body mass. Both nutrient intake and energy expenditure are regulated by a complex interaction between the periphery and the central nervous system. Although not all aspects of central-peripheral interactions involved in energy balance are understood, key factors have been identified. For example, leptin from the adipocyte, ghrelin from the stomach, peptide YY from the gut, and insulin from the pancreas are all involved in the central regulation of energy balance. In the brain, more than a dozen peptides have been implicated in appetite and satiety. Among these peptides, neuropeptide Y, melanocyte-stimulating hormone, and agouti-related peptide in the arcuate nucleus, as well as melanin-concentrating hormone in the lateral hypothalamus, have emerged as important regulators (Fig. 31.2).
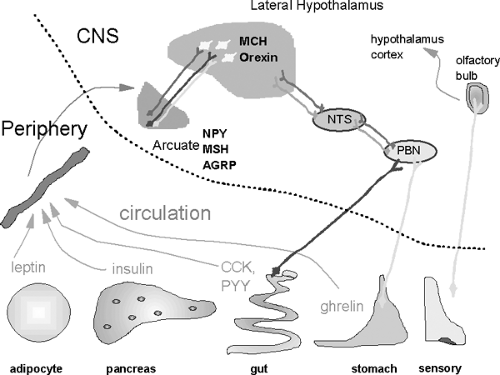 Figure 31.2. Factors in the periphery that impact energy homeostasis are from a variety of tissues and act in the central nervous system (CNS). Peptides known to be important for energy homeostasis include melanocyte-stimulating hormone (MSH), melanocyte-concentrating hormone (MCH), neuropeptide Y (NPY), agouti-related peptide (AGRP). and cocaine-amphetamine-related transcript (CART). Many other central peptides may also play a role in energy balance (orexin, corticotropin-releasing factor, urocortin, galanin, and neurotensin, among others); however, their roles are less well understood. Somatosensory inputs such as smell are also known to be important. CCK, cholecystokinin; PYY, peptide YY. |
It is clear that in mammals the energy-balance equation tips readily toward the overconsumption of calories. The relative threat of starvation to survival apparently has exerted greater evolutionary influence than the long-term consequences of obesity. Indeed, in the wild, the most common cause of death among mice is starvation, as these animals cannot sustain themselves for longer than 3 to 4 days without food. In humans, one excess pound of fat will provide 3,500 calories, which represents adequate fuel for 2 to 3 days in the absence of any food intake.
Role of the Adipocyte in the Regulation of Food Intake
The adipocyte plays an important role in energy homeostasis. To compensate for fluctuations in the availability of food, mammals consume more calories than immediately required for metabolic needs and store excess calories. Calories may be stored as glycogen in the liver, triglycerides in adipocytes (particularly white adipose tissue), and protein in muscle. Adipocyte physiology is regulated by a number of signals, including nutrient availability, hormones, and neuronal input. For example, during a fast, levels of glucose and insulin fall, whereas those of glucocorticoids and growth hormone rise; in consequence, adipocyte triglycerides are metabolized to fatty acids and released into the circulation.
Although this role of the adipocyte in metabolic homeostasis has long been known, its role as an endocrine cell has only recently been recognized. Data suggesting that the adipocyte functions as more than a passive, externally regulated site for the storage of energy date back 10 to 15 years to the description of secretory products such as adipsin and angiotensinogen. Adipsin, a serine protease, is secreted by adipocytes and was found to be markedly decreased in some obese models such as
the ob/ob mouse (42). Nutritionally regulated angiotensinogen secretion also was reported (43). However, the role of the adipocyte as a secretory cell was not fully appreciated until the discovery of leptin, a 16-kDa protein of the cytokine gene family, through genetic analysis of the ob/ob mouse (44). Since the discovery of the ob gene, adipocytes have been recognized to synthesize other factors that may contribute to energy balance.
the ob/ob mouse (42). Nutritionally regulated angiotensinogen secretion also was reported (43). However, the role of the adipocyte as a secretory cell was not fully appreciated until the discovery of leptin, a 16-kDa protein of the cytokine gene family, through genetic analysis of the ob/ob mouse (44). Since the discovery of the ob gene, adipocytes have been recognized to synthesize other factors that may contribute to energy balance.
LEPTIN
A spontaneous mutation in the leptin gene is associated with morbid obesity, hyperphagia, insulin resistance, and infertility in mice. Intriguing studies involving parabiotic mouse pairs had suggested that the syndrome involved the absence of a circulating factor (45,46). Identification of leptin confirmed that absence of a hormone, made in adipocytes and secreted into the circulation, caused the obesity syndrome and that replacement of this factor led to correction of the phenotype (47). Subsequent studies demonstrated an important action of leptin in the hypothalamus (48). The critical importance of leptin in humans was confirmed in studies of morbidly obese children lacking functional leptin alleles. These children, unable to make leptin (49), demonstrate continuous hyperphagia and respond to exogenously administered leptin with a resolution of hyperphagia and significant reductions in body weight (50).
Although the complete absence of leptin is associated with morbid obesity in rare examples of rodent and human obesity, most obese mammals, including humans, have high levels of circulating leptin. Circulating leptin levels correlate well with available fat stores (51), and administration of leptin in most obese states does not lead to decreases in appetite. These findings suggest that the dominant physiologic role of leptin is that of a “starvation signal,” which is important in switching between fed and fasted states rather than in serving as an antiobesity hormone (37,52). In rodents, decreased leptin during fasting is associated with a series of metabolic changes that result in the suppression of reproductive hormones, growth hormone, and thyroid hormones and in the activation of the hypothalamic-pituitary axis with a resultant rise in corticosterone level. These changes can be mitigated by the administration of leptin during the fast. Furthermore, in mice, fasting is associated with a disruption of the estrous cycle, an effect that lasts for many days after the re-introduction of food. Administration of leptin during the fast attenuates this disruption. The physiologic dose response to leptin may be viewed as biphasic. Between absent and normal leptin levels, leptin is effective in signaling adequacy of fat stores, and leptin levels correlate with fat stores. However, when leptin concentrations rise above those associated with adequate adipose stores, leptin has little effect in limiting food intake and a state of “leptin resistance” develops.
ADIPONECTIN
Adiponectin (Acrp30) is a protein exclusively and abundantly expressed in adipose tissue. In mice, a single injection of adiponectin leads to a decrease in glucose levels, and in ob/ob mice, adiponectin abolishes hyperglycemia. In addition, treatment with adiponectin leads to an acute increase in fatty acid oxidation in muscle (53). In isolated hepatocytes, adiponectin leads to a reduction in the amount of insulin needed to suppress gluconeogenesis (54). In various forms of obesity, in both humans and mice, levels of adiponectin messenger RNA (mRNA) are decreased. At least one study has reported an increase in adiponectin after surgery for obesity (55). A recent study indicates that levels are genetically determined and may be associated with obesity (56). These data indicate that adiponectin is important in energy homeostasis and insulin sensitivity.
11-β HYDROXYSTEROID DEHYDROGENASE TYPE 1
The enzyme 11-β hydroxysteroid dehydrogenase type 1 (11-β HSD-1) plays an important role in determining intracellular glucocorticoid concentrations by regenerating active glucocorticoids from inactive cortisone. Activity of this enzyme is relatively increased in visceral fat as opposed to subcutaneous fat (57). Overexpression of the enzyme with a fat-specific transgene leads to a syndrome of visceral obesity that is associated with hyperphagia, insulin resistance, and hypertension (58). The activity of 11-β HSD-1 may be increased in humans with obesity.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


