Nonmalignant Diseases
Benign diseases generally include a class of localized tumors or growths that have a low potential for progression and do not invade surrounding tissue or metastasize to distant sites. Pathologically, they are composed of well-differentiated cells that are considered nonmalignant and usually do not require any treatment. However, clinically, not all benign diseases have benign consequences. Some untreated benign diseases can produce bothersome mass or secretory effects. Others can be locally aggressive and cause secondary debilitating symptoms. For example, Graves ophthalmopathy can lead to local pain and visual impairment without therapeutic intervention1; a hormonally active pituitary adenoma may cause growth abnormality in addition to blindness2; desmoid tumors, can be locally persistent even after surgical resection and some are therefore managed aggressively, similar to their malignant counterparts and may require adjuvant radiation therapy after radical resection.3
Documented empirical use of radiation in imaging and the treatment of benign diseases or conditions occurred soon after the discovery of x-rays by Wilhelm Röntgen in 1895.4 An estimate of over 1 million Americans, mostly young adults and children, received x-ray treatments to the head and neck region for benign conditions between 1920 and 1960.5,6 The painless x-ray treatment and its visible efficacy led to many benign conditions being treated with radiation, such as acne, body hair, scalp ringworm, enlarged tonsils, enlarged thymus, enlarged lymph neck nodes, whooping cough, and others. Radiation therapy was used in some instances due to a lack of effective alternative therapies.7
Over the past decades, advances in medical and surgical therapies have provided new treatment options for many diseases. With improved awareness of late radiation sequelae on normal tissue, particularly radiation carcinogenesis, there has been a gradual decline in the use of radiation therapy for treatment of benign conditions. However, with modern radiation therapy techniques and better understanding of radiobiology, judicial use of radiation still provides good local control in and relief of associated symptoms from a variety of benign diseases.
 RADIOBIOLOGICAL EFFECTS ON BENIGN DISEASES
RADIOBIOLOGICAL EFFECTS ON BENIGN DISEASES
The precise radiobiological mechanisms of radiation effects on benign diseases are not well defined. Radiation is believed to work through a complex of multicellular interactions that affect different cell types in our body system.8 Specific cellular and functional mechanisms depend on the specific disease and site. Although most benign lesions have no known stimuli or causes, some benign lesions may be triggered by trauma as seen in keloid formation after body piercing or heterotopic bone formation after surgery. In conditions that arise following trauma, local inflammation and repair occur, which is often characterized by stimulation of growth factors and accelerated cellular proliferation. For example, in the development of keloids, fibroblast proliferation is responsible for most of the hyperproliferative process. Even with the lower doses commonly used in benign diseases, radiotherapy is clinically effective in inhibiting cell proliferation and suppressing cell differentiation without inducing cell death, as is typically seen with tumoricidal doses of radiation. Yet, radiation can induce apoptosis in selected target cells by influencing the expression of cytokines in macrophages, leukocytes, endothelial, and other cells and thereby modulating the inflammatory cascade.
Among the major sites of radiation effects are the blood vessels; vascular endothelial cells respond rapidly to radiation damage by up-regulating the cytokine-mediated cellular reactions responsible for inflammatory tissue response. Low-dose irradiation (<12 Gy) exerts anti-inflammatory effects on the endothelial cells of capillaries and mononuclear cells of the immune system.9
Cell adhesion molecules, selectins, are mobilized to the cell membrane and change the capillary permeability, allowing the inflammatory cells (lymphocytes, macrophages, monocytes) to migrate into interstitial space. The anti-inflammatory effect is attributed to the modulation of cytokine and adhesion molecule expression on the activated endothelial cells and leukocytes. These cells are known to be radiosensitive. They express proinflammatory cytokines (e.g., interleukin-1, interleukin-6) or necrosis factors (e.g., tumor necrosis factor-α), which influence the complement cascade and enzymes of inflammatory reaction. Interleukin-1 stimulates the production and release of proinflammatory prostaglandins, leading to a change in synthesis of inducible nitric oxide synthetase.
The radiation-induced modulation of nitric oxide production and oxidative burst in activated macrophages and native granulocytes lead to modification of the immune response and inflammatory process as well as clinical analgesic effects. Although endothelial cells possess a high proliferative potential and are sensitive to radiation damage at high doses, they are not prone to rapid mitotic radiation death at low doses.
Chronic inflammatory processes are triggered by antigen–antibody reactions and mediated by mononuclear peripheral blood cells in the immune system. Ionizing radiation helps suppress some of these cell populations, such as T-lymphocytes, in the inflammation process or modulate their effects. Although low doses of radiation can exert an anti-inflammatory response, high doses of radiation as used in malignant tumors can elicit proinflammatory effects and fibrotic change in normal tissue.10 At higher single or total doses, endothelial cell damage can lead to sclerosis and obliteration of blood vessels. In vascular disorders such as hemangiomas or arteriovenous malformations, high radiation doses may induce occlusion of pathologic vessels. In addition to inhibition of cell proliferation, cell killing may play a part in the management of benign meningiomas, pituitary adenomas, or neuromas where higher, tumoricidal doses of radiation may be required.
 RISK OF SECOND MALIGNANCIES
RISK OF SECOND MALIGNANCIES
The induction of cancer or genetic defects by radiation exposure is attributed to stochastic effects where there is no threshold level of radiation exposure below which cancer induction or genetic effects will not occur. Increasing the radiation dose or the volume of exposure will increase the probability that a cancer or genetic effect will occur. Sometimes, the radiation effects are difficult to separate from inherent genetic effects. For example, in patients with retinoblastoma, the Rb1 gene plays an important role in the development of radiation-induced sarcomas.11,12 In a study of 384 retinoblastoma patients treated with radiation, the actuarial risk for developing a sarcoma in the treatment field 18 years after treatment was 6.6%.11 In another study of 693 patients, the cumulative risk for any sarcoma 50 years after radiotherapy was 13.1%.12 Although most sarcomas were within the irradiated fields, 18 of 69 sarcomas developed outside the treatment fields. Rb1 mutations appear to confer a genetic predisposition to developing sarcomas, especially after radiation exposure.
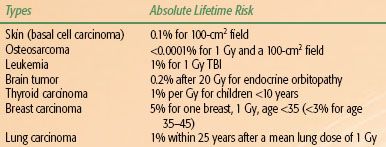
The risk of the induction of secondary tumors was overestimated in the past.13 Trott and Kamprad14 used the epidemiologic data from long-term follow-up studies on patients treated with radiotherapy for benign diseases to estimate the risk of cancer induction. Taking all known modifying and organ-specific factors into account, including doses of radiation and volume irradiated, the estimated absolute lifetime risk for sarcoma induction was <0.0001% for 1 Gy and a 100-cm2 field. Table 91.1 lists the absolute lifetime risk for other malignancies.
Jansen et al.15 applied the effective dose concept and estimated the carcinogenic risk in patients after radiotherapy of benign diseases (heterotopic ossification, omarthritis, gonarthrosis, heel spurs, and hidradenitis suppurativa). Special risk modifying factors, including age at exposure and gender, were taken into account. For an average-aged population, the estimated number of radiation-induced fatal tumors was between 0.5 and 40 persons per 1,000 patients treated. The range of effective doses was also found to be large (5 to 400 mSv). In addition to age and gender, the individual risk also depends on individual inherent sensitivity, anatomic site, type of disease, and treatment technique, such as dose and fractionation.
TABLE 91.1 THE ESTIMATED ABSOLUTE LIFETIME RISK FOR MALIGNANCIES AFTER RADIATION THERAPY FOR BENIGN DISEASES
 INDICATION FOR RADIOTHERAPY
INDICATION FOR RADIOTHERAPY
The majority of benign diseases can be classified as inflammatory, degenerative, hyperproliferative, or functional. Therefore, therapeutic approaches vary widely and are regionally customized, in part because of geographic traditions and differences in clinical training. Radiation treatment of benign diseases is less commonly used in the United States than in other parts of the world where variation in indications and treatment schedules are institutionally based.16 Within Germany, a pattern of care study revealed significant geographic and institutional differences.17 Although most radiation treatments for benign disease are delivered in the low-dose range (<10 to 15 Gy), the prescribed dose varied widely and inconsistently within geographic regions and among institutions.
Degenerative processes in tendons, ligaments, and joints can cause pain by chronic inflammation and trigger secondary functional impairment of the involved musculoskeletal system. Although radiation does not halt the degenerative process, it may reduce the inflammation and provide partial or complete pain relief. This clinical effect is well established in reports of osteoarthritis, synovitis, and bursitis, where low-dose radiation therapy has improved the function of affected joints.17,18
Benign diseases may have a significant effect on self-image and self-esteem because of cosmetic appearance (e.g., facial keloids, juvenile angiofibroma) or have a lasting impact on quality of life because of chronic pain or other secondary symptoms (e.g., heterotopic bone, macular degeneration). When benign diseases become locally invasive with aggressive growth, therapeutic intervention can prevent or limit functional loss of organs. In rare cases of large hemangioma with associated thrombocytopenia and consumption coagulopathy (Kasabach-Merritt syndrome), potentially fatal complications can occur, and timely therapeutic intervention can be life-saving.19
Although there is a lack of international consensus, the German Working Group on Radiotherapy of Benign Diseases published their consensus guidelines for radiation therapy of nonmalignant diseases. The guidelines were to serve as a starting point for quality assessment, prospective clinical trials, and outcomes research.18 In brief, treatment is indicated when benign diseases are symptomatic or potentially symptomatic. When other methods are unavailable or have failed, radiation therapy should be considered. As medical professionals, we remain mindful of therapeutic gain and potential treatment side effects and complications. A thorough risk–benefit analysis is always pertinent. Organ-specific acute and chronic toxicities, including potential effects on fertility and induction of secondary tumors in the future, must be explained to and discussed with patients, especially those who are young and have a long life-expectancy. Informed consent, which is required for all medical interventions, is certainly required for treatment of benign diseases and should be obtained prior to the delivery of radiation therapy.
This chapter covers some of the more common benign conditions that are still encountered in the practice of radiation oncology. Details on the therapeutic approaches and data on radiation dose regimens for different benign diseases are summarized in the individual corresponding sections.
 BENIGN NEOPLASMS OF THE BRAIN, HEAD, AND NECK
BENIGN NEOPLASMS OF THE BRAIN, HEAD, AND NECK
Nonmalignant tumors of the central nervous system (CNS) and neck can lead to severe, life-threatening symptoms due to pressure and mass effect on critical structures from tumor growth. However, depending on tumor growth rate and location, the surrounding tissue may adapt, leading to a delay in the clinical diagnosis.
Meningioma
Background and Clinical Aspects
Meningiomas are the most common benign tumors of the CNS. The incidence peaks in the seventh decade of life with a 2 to 1 female-to-male predominance. The majority (>90%) of meningiomas are benign and classified by the World Health Organization (WHO) as grade I tumors.20 WHO grade II meningiomas (atypical, clear cell, or chordoid) have a higher tendency for local recurrence, and WHO grade III malignant meningiomas (anaplastic, rhabdoid, papillary) are exceedingly rare.
The most common presenting symptom is headache, but patients may present with other localizing symptoms depending on the tumor location. The radiographic diagnosis of meningioma is often made on computed tomography (CT) or magnetic resonance imaging (MRI) based on the appearance of a homogeneously and intensely enhancing extra-axial mass with or without the presence of a dural tail.
Surgical Management
Surgical resection is the treatment of choice for the majority of patients, as this will relieve symptoms and also provide a pathologic diagnosis. The primary goal of surgery is to remove as much tumor burden as possible while minimizing the risk of neurologic deficits (maximal safe resection). Gross total resection (GTR) is generally attempted for patients with tumors in locations such as the convexity and olfactory groove.21,22 After GTR, the relapse rate is as low as 10%, but this depends on the Simpson classification, which grades tumors according to extent of resection and degree of dural involvement (Table 91.2).23 Local recurrence rates are as high as 40% for patients with incomplete resection,23 although these rates can be substantially reduced with the use of adjuvant radiotherapy.
Meningiomas tend to be highly vascularized tumors. In select patients, preoperative embolization is used to decrease blood loss and improve the extent of resection.24,25
Active Surveillance
Asymptomatic patients with small meningiomas may be observed clinically. At the time of tumor growth or the development of symptoms, patients can be treated with surgery or radiation therapy. The safety and reasoning for this approach was established in a large retrospective series from Japan that demonstrated that the majority of patients do not require intervention in the short term.26
TABLE 91.2 SIMPSON GRADING SYSTEM FOR POSTOPERATIVE MENINGIOMAS WITH ASSOCIATED RATES OF RECURRENCE

Systemic Therapy
Interest in the use of medical therapy to treat meningiomas stems from the observation that up to 67% of meningiomas express the progesterone receptor or androgen receptor, and approximately 10% express the estrogen receptor.27 However, response rates to antihormonal agents are low. Overall, studies that have investigated the role of chemotherapy, such as hydroxyurea, in the management of recurrent disease have demonstrated little efficacy.27
Radiotherapy
Primary radiotherapy (RT) is indicated for tumors in locations in which complete resection is not feasible (i.e., optic nerve, cavernous sinus, major venous sinus) or for patients who are poor surgical candidates. Adjuvant RT is indicated for patients with subtotal resection (STR), recurrent disease, or for WHO grade II or III tumors. RT techniques include conventionally fractionated three-dimensional conformal radiotherapy (3D-CRT), conventionally fractionated intensity-modulated radiation therapy (IMRT), frame-based or linear accelerator–based fractionated stereotactic radiotherapy (FSRT), stereotactic radiosurgery (SRS), or protons and heavy ions.
The MRI sequences that best delineate the gross tumor volume (GTV) should be coregistered with the treatment-planning CT scan for optimal treatment planning and delivery. Particularly for patients receiving FSRT or SRS, it is important that a neuroradiologist and neurosurgeon be involved in assisting with GTV delineation, as enhancement from residual tumor versus postoperative change is often difficult to ascertain.
For 3D-CRT or IMRT treatments, the clinical target volume (CTV) is constructed by adding a 1- or 2-cm symmetric margin around the GTV, respecting normal tissue boundaries. An additional 3 to 5 mm is added for the final planning target volume (PTV). These margins may be modified based on institutional policy and other considerations, such as the availability of daily image guidance (i.e., kilovolt imaging or cone-beam CT).
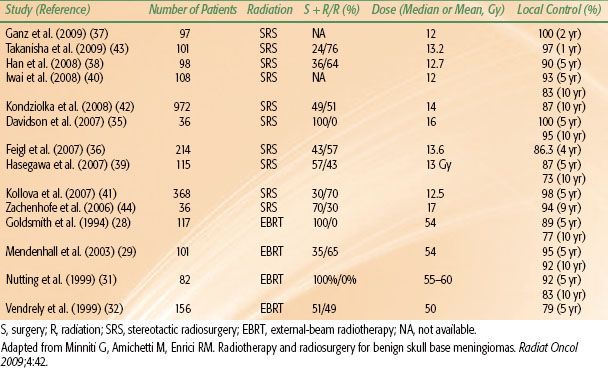
For benign meningiomas, the typical dose prescription to the PTV is 50 to 54 Gy given in 1.8- to 2-Gy daily fractions. Retrospective data suggest that local control is inferior for patients treated with doses of <52 Gy.28 For patients with more aggressive histology (WHO grade II or III tumors), the GTV is expanded by at least 2 cm, with a higher dose prescription in the range of 59.4 to 63 Gy. Several modern series of radiotherapy show 5-year local control rates ranging from 89% to 98%, with three-dimensional conformal therapy demonstrating local control rates >95% (Table 91.2).28–30,31–32
Because meningiomas are frequently noninvasive and well-circumscribed tumors, SRS and FSRT are increasingly being used in their treatment. The decision to fractionate depends largely on tumor size and proximity to critical structures, such as the optic apparatus or brainstem. Typical dose prescriptions for frame-based SRS range from 12 to 16 Gy prescribed to the 50% isodose line (IDL) and 14 to 18 Gy prescribed to the 80% IDL for a frameless robotic radiosurgery platform. In patients with tumors that require fractionated treatment, dose prescriptions vary and are dependent on the individual case. For example, at Stanford University, primary or residual meningiomas of the convexity and skull base are treated with 15 to 18 Gy in 1 or 2 fractions. Additionally, a select group of perioptic tumors, including meningiomas, have been treated with a prescription of 24 to 30 Gy in 3 to 5 fractions (to the 80% IDL) with high rates of tumor control and visual preservation (Fig. 91.1).33 Recent nonrandomized, prospective evidence indicates that FSRT should be the treatment of choice for optic nerve sheath meningiomas due to the high rate of preservation of visual acuity.34
Reported results with SRS are excellent, with 5-year local control rates as high as 98% to 100% (Table 91.3).35–41,42,43,44 DiBiase et al.45 demonstrated that male gender, conformality index <1.4, and size >10 mL predict for worse outcome after SRS. That article also showed improved disease-free survival in patients in which the dural tail was covered as part of the target volume. The benefit of including the dural tail has to be weighed against the risk of toxicity from increasing the target volume for each individual case.
Due to their physical properties, protons and heavy ions (i.e., carbon) are attractive choices for the treatment of meningiomas, particularly for those located near critical structures. Several studies have shown excellent local control rates with the combination of protons and photons or protons alone.46–49 In the study by Weber et al.,48 patients were treated to a median dose of 56 cobalt gray equivalents (GyE) given in 1.8 to 2.0 GyE per day.
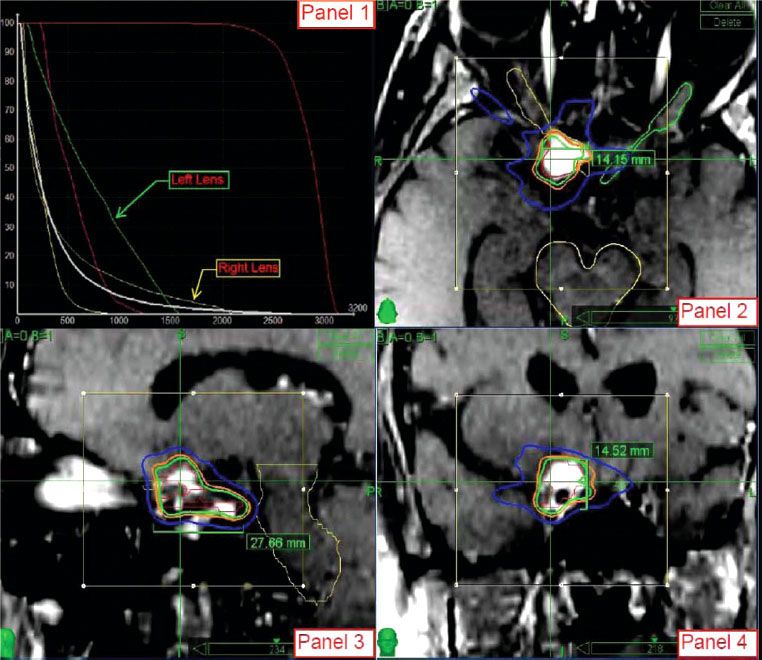
FIGURE 91.1. Radiosurgery treatment plan of a patient with a right optic nerve sheath meningioma treated to a dose of 24 Gy in 3 fractions. The lesion is intensely enhancing on the postcontrast stereotactic magnetic resonance imaging sequences. Panel 1 demonstrates the dose–volume histogram for the patient. The maximum dose to the ipsilateral optic nerve was 22.3 Gy. Panels 2–4 demonstrate the isodose curves for the treatment in the axial, sagittal, and coronal planes, respectively. The 100% (24 Gy) isodose line is green, the 88% (21 Gy) isodose line is orange, and the 50% (12 Gy) isodose line is blue.
Pituitary Adenoma
Background and Clinical Aspects
Pituitary adenomas comprise 10% to 15% of all intracranial tumors. Approximately 75% of these tumors are functional (secretory), thereby producing increased amounts of hormones. Prolactinomas and growth-hormone (GH)–secreting adenomas are the most frequently encountered. Functional adenomas are more common in women, while nonfunctioning and GH-secreting adenomas are more common in men.
Adenomas are often classified by size, with a picoadenoma <0.3 cm, microadenoma <1 cm, and macroadenoma >1cm. Macroadenomas may exert mass effect upon the optic chiasm, leading to the classic sign of bitemporal hemianopsia. Headaches are seen in approximately 20% of patients. If the adenoma extends to the cavernous sinus, cranial nerve deficits may be present. Involvement of the hypothalamus by the adenoma results in hypopituitarism.
Patients with functional adenomas present with signs and symptoms that correspond to the excess hormone: galactorrhea, amenorrhea, diminished libido, and infertility in patients with prolactinomas; acromegaly or gigantism in patients with GH-secreting adenomas; Cushing disease in adrenocorticotropic hormone (ACTH)–secreting adenomas; hyperthyroidism in patients with thyroid-stimulating hormone (TSH)–secreting adenomas. In patients who have had bilateral adrenalectomy, up to 40% will develop Nelson syndrome, which is characterized by an ACTH-secreting adenoma and increased skin pigmentation secondary to increased release of α-melanocyte–stimulating hormone.
In addition to history and detailed physical examination (H&P), workup of a pituitary tumor includes laboratory analysis of pituitary hormone levels, contrast enhanced MRI with thin slices through the pituitary (Fig. 91.2A,B), and tissue diagnosis to rule out other causes of pituitary masses, including craniopharyngioma, meningioma, suprasellar germ cell tumor, metastatic disease, or a benign lesion (i.e., cyst).
TABLE 91.3 CLINICAL OUTCOMES OF STEREOTACTIC RADIOSURGERY OR EXTERNAL-BEAM RADIOTHERAPY (WITH OR WITHOUT SURGERY) FOR MENINGIOMAS IN MODERN SERIES
Surgical Management
Surgery is generally the treatment of choice for pituitary adenomas. Surgery provides immediate relief of compressive symptoms and helps to decrease hormone secretion. The most common surgical technique is through a transsphenoidal approach. In some cases, a more aggressive surgery (i.e., frontal craniotomy) may be indicated for patients with extensive intracranial and skull-based involvement. Overall, local control rates range from 50% to 80% after surgery alone for both functioning and nonfunctioning adenomas.50 In patients who continue to have abnormally elevated hormones after surgical resection, adjuvant treatment with pharmacotherapy or radiation therapy is pursued.
Pharmacotherapy
Pharmacotherapy, such as bromocriptine and cabergoline for prolactinomas, octreotide for GH adenomas and TSH adenomas, and ketoconazole for ACTH adenomas, is often used as an adjunct to surgery for patients with functioning adenomas. With the exception of prolactinomas, the use of these drugs as monotherapy is generally not curative. Prolactinomas can often be managed with pharmacotherapy alone, but a high proportion of patients are unable to tolerate bromocriptine for long periods of time due to nausea, headache, and fatigue.
Radiotherapy
Except for medically inoperable patients in which RT is used in the primary setting, the role of RT is generally in the adjuvant setting with the following indications: recurrent tumor after surgery; persistence of hormone elevation after surgery; residual disease after STR or debulking procedure. Tumor growth control is excellent, particularly for patients with nonfunctioning adenomas.51,52–53 Endocrine control after treatment of functioning adenomas, as demonstrated by normalization of pituitary hormone levels, takes years to develop. At a median of 2 years after RT, growth hormone levels stabilize quickest; normalization is slowest for TSH-secreting adenomas.54 Pharmacologic therapy should be discontinued 1 to 2 months prior to the initiation of RT based on evidence demonstrating lower RT sensitivity with concurrent medical treatment.55
RT techniques include 3D-CRT, IMRT, single-fraction SRS, and FSRT. Delineation of the GTV (or preoperative GTV in the case of GTR) should be performed by coregistration of the postoperative MRI to the treatment planning CT scan.
For 3D-CRT and IMRT, the CTV is constructed by adding 1 to 1.5 cm to the GTV; an additional 3 to 5 mm is added to the CTV to create the PTV. These margins may be modified based on institutional policy and other considerations, such as the availability of daily image guidance. Nonfunctional adenomas are typically prescribed a dose of 45 to 50.4 Gy given in 1.8- to 2.0-Gy daily fractions (Fig. 91.2C–E). Higher doses in the range of 50.4 to 54 Gy are recommended for secretory adenomas.
SRS remains an attractive option for the treatment of pituitary adenomas. General principles apply in that FSRT is used over SRS for large lesions (>3 cm) or lesions near critical structures (<1 or 2 mm from the chiasm). Similar to 3D-CRT or IMRT, higher doses are needed for functional adenomas compared with nonfunctional adenomas. Numerous retrospective studies have demonstrated excellent local control rates of 92% to 100% for nonfunctional adenomas, using doses of 14 to 25 Gy (at the edge of the tumor) in a single fraction.56 Commonly used prescriptions are 16 to 20 Gy in a single fraction for nonfunctional adenomas and 20 to 25 Gy in a single fraction for functional adenomas using a frameless robotic radiosurgery platform.
Craniopharyngioma
Background and Clinical Aspects
Craniopharyngiomas comprise 6% to 10% of pediatric CNS tumors, or approximately 300 to 350 cases per year in the United States. The median age of diagnosis is 5 to 10 years, with a second peak in patients >40 years old.
These benign tumors are epithelial, arising from remnants of Rathke’s pouch (hypophyseal-pharyngeal duct), and are most commonly located in the suprasellar region, although they may be found in the sella proper. Craniopharyngiomas generally abut the hypothalamus and third ventricle. Histologically, they are divided into the adamantinomatous and squamous subtypes. The adamantinomatous subtype is characterized by a solid and cystic pattern with the well-known description of “machine oil-like” cystic fluid.
FIGURE 91.2. A recurrent nonfunctioning pituitary adenoma 7 years after surgical resection in the axial (A) and coronal (B) planes. The yellow arrows denote invasion into the left cavernous sinus. Rapid arc intensity-modulated radiotherapy treatment plan for the same patient in the axial (C), coronal (D), and sagittal (E) planes. The planning target volume (purple shaded area) was prescribed at 50.4 Gy in 28 fractions.
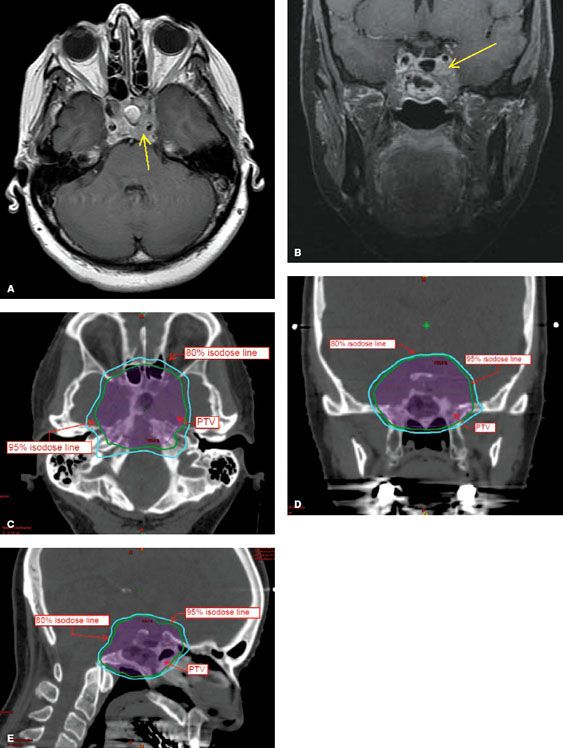
FIGURE 91.3. Axial, coronal, and sagittal magnetic resonance imaging of a patient with multicystic (yellow arrows) craniopharyngioma prior to treatment.
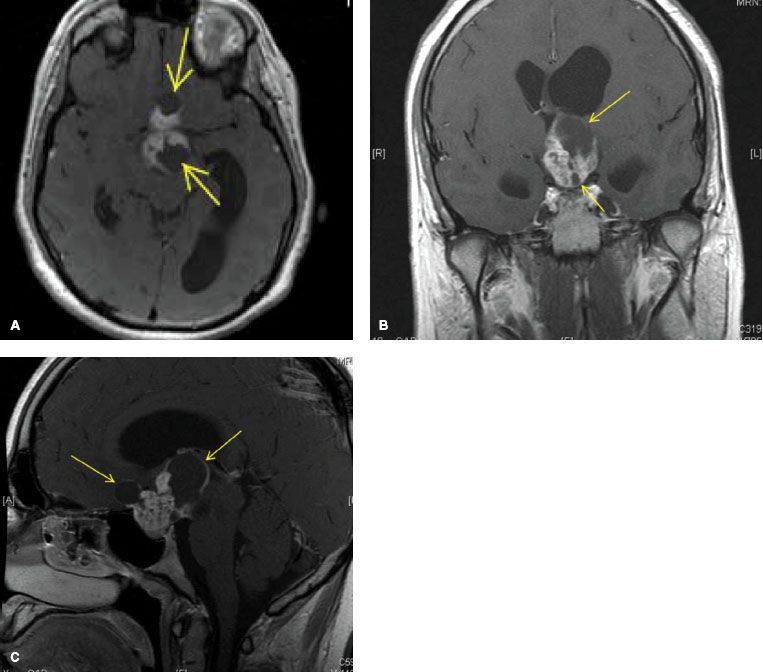
Presenting signs and symptoms include headache, nausea and vomiting, bitemporal hemianopsia, and endocrine dysfunction (diabetes insipidus, dwarfism, fat tissue disturbance, adrenal cortical insufficiency). The most common hormone deficiency is lack of GH. The workup is similar to that of pituitary adenoma and includes H&P, pituitary hormone levels, and brain MRI with thin slices through the sella (Fig. 91.3).
Surgery
The primary goal of surgery is complete resection. However, GTR may be associated with high rates of neurologic sequelae, including visual impairment and panhypopituitarism. In order to minimize morbidity, most patients are treated with maximal safe resection followed by adjuvant RT. In some cases, intralesional bleomycin may be directly injected into the cyst to decrease the rate of cyst recurrence.
Radiotherapy
Radiation therapy is often used in the adjuvant setting. In select patients (i.e., <3 years old), observation following STR may be an option as local control rates are similar with RT at the time of relapse (“salvage” RT) compared with adjuvant RT with no compromise in overall survival.57
RT techniques include 3D-CRT, IMRT, FSRT, proton therapy, and intralesional RT with β-emitting isotopes (yttrium-90, phosphorous-32). The GTV is the postoperative residual tumor volume, including the cyst wall, if present. The postoperative MRI should be fused with the treatment planning CT scan for optimal target delineation. A margin of 1 to 1.5 cm is added to the GTV to create the PTV. Dose prescriptions for 3D-CRT and IMRT are typically 54 Gy given in 1.8-Gy daily fractions.
Fractionated proton radiotherapy has demonstrated excellent results. The Loma Linda series treated 15 patients to a total dose of 50.4 to 59.4 GyE given in 1.8-GyE daily fractions.58 Local control was achieved in 14 of 15 patients, with few long-term complications. In a series from the Massachusetts General Hospital (MGH), no failures were seen in 24 patients who received fractionated proton radiotherapy to a total dose of 52.2 to 54 GyE in 1.8 GyE per fraction.59
It has been well established that cysts may regrow during the several weeks of fractionated treatment. The MGH proton study recommends that reimaging (CT, or MRI if the cyst is not well visualized on CT) be performed within 2 weeks of the treatment planning scan and every 2 weeks thereafter; for large cysts or those that demonstrate growth during RT, weekly reimaging is recommended.59 The emergence of image-guided radiotherapy techniques now allows for the convenient monitoring of cyst regrowth with cone-beam CT scans while patients are on the treatment table.
SRS and FSRT have been used with success in the treatment of craniopharyngioma. In one series from Stanford University using a frameless robotic platform,60 16 patients were treated postoperatively with doses of 18 to 38 Gy given over 3 to 10 fractions prescribed to mean IDL of 75%. Local control was 91% in this cohort of patients with no visual or neuroendocrine complications. Similar results have been demonstrated with use of a frame-based platform.61–64
Cystic craniopharyngiomas may also be managed by the use of intralesional radioactive isotope injection using a β-emitter. Typical prescriptions range from 200 to 250 Gy prescribed to the cyst wall. Optimal results are seen in patients whose tumors have one cyst and lack a large solid component.65,66
Acoustic Neuroma (Vestibular Schwannoma)
Background and Clinical Aspects
Acoustic neuromas (AN) represent 5% to 8% of primary CNS brain tumors. They are derived from Schwann cells of the neurilemma of the vestibulocochlear nerve (CN VIII). The vast majority of cases (90%) are unilateral and sporadic. Bilateral AN occur in about 10% of cases and are associated with the autosomal dominant disorder neurofibromatosis type II.
Symptoms include sensorineural hearing loss, tinnitus, and vertigo. Hearing loss is correlated more with tumor location (intracanalicular) rather than tumor size. In a minority of patients (5%), facial nerve symptoms may be present. As the AN grows, it may affect the trigeminal nerve (CN V) and brainstem.
During the initial workup, physical examination should include the Rinne test (air conduction greater than bone conduction on the affected side) and Weber test (vibratory sound louder on the unaffected side) to test for sensorineural hearing loss, as well as detailed examination of CN V and CN VII. All patients should undergo audiometry when the diagnosis of AN is suspected; this will often reveal asymmetric hearing loss more prominent at high frequencies as well as impairments in speech discrimination score. Imaging should include a contrast-enhanced MRI with thin slices through the internal auditory canal. The entire neuraxis should be imaged in patients with neurofibromatosis type II.
Surgery
For many years, the standard treatment for patients with AN was microsurgical resection, either via a translabyrinthine approach or middle cranial fossa approach. Surgery remains the preferred treatment for patients with large, symptomatic lesions. Hearing preservation is approximately 50% to 60% after surgery, and facial nerve preservation ranges from 80% to 90%.67,68
Active Surveillance
Observation is appropriate management for asymptomatic patients with small tumors. Serial MRI and audiometry (at least once per year) should be performed in this patient cohort for surveillance. Treatment is initiated when the lesion demonstrates rapid and significant growth or when the patient becomes symptomatic.
Radiotherapy
RT in the form of SRS or FSRT is an option for the primary treatment of AN, often with higher rates of hearing preservation and facial nerve presentation compared with surgery. Proton therapy has also been used to treat AN.
SRS doses using frame-based platforms are generally 12 to 13 Gy prescribed to the 50% IDL. Although earlier studies demonstrated lower hearing preservation rates with higher SRS doses,69 current results are significantly improved. Flickinger et al.70 demonstrated a local control rate of 98.6%, hearing preservation rate of 70.3%, trigeminal neuropathy rate of 4.4%, and no incidence of facial nerve dysfunction. Using a frameless robotic radiosurgery platform to treat 383 AN to a dose of 18 to 21 Gy given in 3 fractions, Stanford University researchers demonstrated a 98% local control, 76% hearing preservation, 2% incidence of trigeminal nerve dysfunction (transient in four of the eight affected patients), and no facial nerve dysfunction.71
Common dose prescriptions employing FSRT include 25 Gy in 5 fractions, 30 Gy in 10 fractions, and 50 to 55 Gy in 25 to 30 fractions. A nonrandomized prospective trial compared SRS (10 to 12.5 Gy) to FSRT (20 to 25 Gy in 5 fractions). This study demonstrated comparable rates of local control, hearing preservation, and CN V and CN VII preservation between the two groups.72
Proton beam SRS has also been used to treat AN, although with low rates of hearing preservation compared with other RT techniques. In a series from MGH, 88 patients were treated to a median dose of 12 GyE given in a single fraction prescribed to a median IDL of 70%. Tumor control rates at 2 and 5 years were 95.3% and 93.6%, respectively. Facial and trigeminal nerve preservation rates were 90%. Only 33% of patients retained serviceable hearing.73 In a study of FSRT using a proton beam, local control was excellent but the hearing preservation rate was poor at 42%.74
Chordoma
Background and Clinical Aspects
Chordomas are rare, slowly growing midline tumors originating from the embryonal notochord rests in the skull base (35%), vertebral column (15%), or sacral regions (50%). The most common sites of skull-based tumors include the clivus, dorsum sella, and nasopharynx.
Patients typically present with signs and symptoms attributable to the primary site of the tumor. Workup includes MRI with contrast enhancement. CT may complement MRI to assess for local bony destruction. A biopsy is necessary primarily to distinguish chordoma from chondrosarcoma, which has a better prognosis, and other malignancies. In children, biopsy is essential to distinguish chordoma from rhabdomyosarcoma, which can frequently present in the same location.
Surgery
Complete surgical resection is the mainstay of treatment. However, due to location, GTR is often not possible. Relapse rates are as high as 50% even after surgical resection with negative margins.75 Poor prognostic factors include large tumors, recurrent tumors, older age, and presence of necrosis on biopsy.76–77,78
Systemic Therapy
Approximately 25% of chordomas metastasize to the lungs, liver, or bone. In these patients, or in patients with recurrent disease after surgical resection and radiation therapy, molecularly targeted agents may be considered. Several small studies have demonstrated some benefit to the use of imatinib or the combination of imatinib and sirolimus in this situation.79–81
Radiotherapy
Adjuvant radiation therapy is indicated to reduce recurrence rates for skull-based chordomas. Retrospective data suggest that salvage RT is inferior to adjuvant RT with 5- and 10-year overall survival rates of 50% and 0%, respectively, for those treated with salvage RT compared with 80% and 65%, respectively, for patients treated with adjuvant RT.82 RT techniques for treatment of skull-based chordomas include conventionally fractionated 3D-CRT or IMRT, fractionated charged particle therapy (protons, carbon ions), and FSRT.
Determination of the GTV should be made by coregistration of the preoperative and/or postoperative MRI to the treatment planning CT scan. Particularly for pediatric cases in which IMRT will be used with tight margins, a neuroradiologist should be available at the time of treatment planning to assist in creating the GTV. Margins for CTV should be 1 to 2 cm with an additional 3 to 5 mm for the PTV. Dose prescriptions to the PTV for patients receiving photon-based treatment should be at least 60 Gy given in 1.8- to 2.0-Gy daily fractions.
Proton-based therapy can achieve higher doses with good results. In a series of 195 chordomas treated at the MGH, 5-year progression-free survival was 70% with doses of 63 to 79.2 GyE given in 1.8- to 2.0-GyE daily fractions.83 Research at Loma Linda examined 33 cases of chordomas treated with a median of 70 GyE and found a 76% 5-year local control and 79% 5-year overall survival.78
In a report of 96 chordomas treated using carbon ion therapy at the University of Heidelberg, with median total dose of 60 GyE (range 60 to 70 GyE) delivered in 20 fractions within 3 weeks, good local control rates of 81% at 3 years and 70% at 5 years were observed.84 There was a trend to improved local control in patients who received >60 GyE.
FSRT and SRS are less well established than charged-particle therapy. The North American Gamma Knife Consortium published the experience of 71 patients who underwent Gamma Knife (Elekta Corp, Stockholm) SRS as primary, adjuvant, or salvage therapy for skull-base chordomas.85 The median dose to the tumor margin was 15 Gy (range 9 to 25 Gy). Five-year local control was 66% for the entire cohort (69% for the no prior RT group and 62% for the prior RT group). Debus et al.86 reported on 37 patients with chordomas treated with FSRT to a median dose of 66.6 Gy given in 1.8-Gy daily fractions; the target volume was encompassed by 90% of the dose. Local control was 82% at 2 years and 50% at 5 years.
Glomus Tumor, Chemodectoma, and Paraganglioma
Background and Clinical Aspects
Glomus tumors are rare, benign tumors that occur at and along the carotid artery near the bifurcation (carotid body tumor), the jugular bulb (glomus jugulare), or the middle ear (globus tympanicum). The peak age is in the fifth decade of life. Bilateral or multiple tumors occur in 10% to 20% of affected patients.
Symptoms include headache, cranial nerve dysfunction, dysphagia, pulsatile tinnitus, vertigo, and large, pulsating masses in the neck. In rare cases, patients present with episodic hypertension, which may be related to the secretion of vasoactive substances by the tumor. In this situation, urine and serum metanephrines should be measured. The clinical presentation coupled with imaging (high-resolution CT, MRI, or angiography) often establish the diagnosis. In patients in which multiple tumors are suspected, imaging with metaiodobenzylguanidine may be useful.
Surgery
In the carotid region, primary tumor resection after previous embolization is the therapy of choice. At the skull base or tympanum, neurosurgical intervention is often deferred due to the high rates of complications, including stroke and cranial nerve injury.87
Radiotherapy
RT is indicated for patients with tumors in unsuitable locations (i.e., skull base), as adjuvant therapy after STR, or as salvage therapy at the time of relapse after surgery. RT techniques include conventionally fractionated 3D-CRT or IMRT, SRS, and FSRT.
The diagnostic MRI should be coregistered with the treatment planning CT scan. The GTV is delineated and 1 to 1.5 cm is added for clinical and setup margin. With conventional techniques, doses are often 45 to 55 Gy given in 1.8- to 2.0-Gy daily fractions with local control rates near or >90% in several series.88–91
Results with SRS have been comparable to that of conventionally fractionated RT. Using a frame-based platform, reported tumor margin doses range from 12.5 to 20 Gy prescribed to the 50% IDL92,93–94 and 15 to 25 Gy for linac-based SRS.87 Local control rates in these series range from 90% to 100%. A recent meta-analysis of SRS published by the Johns Hopkins Hospital reviewed 335 patients treated with SRS across 19 different studies.95 In the eight studies with median follow-up >3 years, clinical control was 95% and tumor control was 96%. The control rates were equal among patients treated with linac-based platforms and frame-based platforms. Complications were rare and often transient in each of the studies examined. On the basis of these findings, the authors advocate for the use of SRS as primary treatment of glomus jugulare tumors.95
Juvenile Nasopharyngeal Angiofibroma
Background and Clinical Aspects
Juvenile nasopharyngeal angiofibroma (JNA) is a rare, benign, vascularized tumor in the head and neck, affecting mostly male adolescents. JNAs develop from the sphenoethmoidal suture and spread from the nasal cavity to the sphenopalatine foramen and pterygopalatine fossa. Other routes of local spread include the paranasal sinuses, infratemporal fossa, orbital space, and middle cranial fossa.
Symptoms initially include recurrent epistaxis and impaired nose breathing. As local extension occurs, patients may develop facial swelling, orbital symptoms (blindness), cranial nerve deficits, and headaches from intracranial extension.
Diagnosis is often made with the clinical presentation and CT or MRI-based imaging. Biopsy may cause massive bleeding. Numerous staging systems are used to categorize the tumors based on extent of local extension, including the Chandler et al.,96 Fisch,97 and Radkowski et al.98 systems (Table 91.4).
Surgery
Surgery combined with embolization is the preferred treatment. Through surgery, most JNAs without intracranial extension (i.e., Chandler stage I to III) have local control rates of near 100%. In patients with intracranial extension, complete resection is often not possible.
TABLE 91.4 CHANDLER STAGING SYSTEM FOR JUVENILE NASOPHARYNGEAL ANGIOFIBROMAA

TABLE 91.5 CLINICAL RESULTS OF RADIOTHERAPY IN JUVENILE NASOPHARYNGEAL ANGIOFIBROMA
Radiotherapy
Tumors with intracranial extension or tumors in patients who are medically inoperable are generally treated with RT as the primary modality. Indications for postoperative RT include relapse after surgery. Fractionated IMRT is currently the RT technique of choice to limit collateral radiation to critical structures near the target volume.
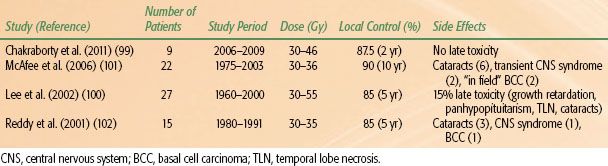
The PTV is generally treated to 30 to 50 Gy given in fraction sizes of 2 to 3 Gy per day. In modern series, local control rates range from 85% to 100% (Table 91.5).99,100,101,102 After RT, JNA remission is slow, and late recurrences may occur.
Langerhans Cell Histiocytosis (Histiocytosis-X)
Background and Clinical Aspects
Langerhans cell histiocytosis (LCH) is a rare disorder that affects approximately 300 individuals with a higher incidence in children (3 to 5 per million) than adults (1 to 2 per million). Age is an important prognostic factor, with children having better outcomes than adults. In the past, it was felt that children younger than 2 years old were felt to have a poor prognosis, but recent data from the LCH-II study now refute that point.103
The disease is due to an accumulation or proliferation of cells that phenotypically resemble the Langerhans skin cell and can cause tissue damage by production of cytokines and infiltration. The actual Langerhans cell is a myeloid dendritic cell that expresses the same antigens (CD1a and CD207) as the Langerhans skin cell.104 On electron microscopy, Birbeck granules are the classic finding.
LCH can affect a variety of organ systems. Patients are typically stratified into groups based on the extent of disease: single-system disease at a single site, single-system disease involving multiple sites, or multisystem disease. The clinical presentation is dependent on the sites of disease. Involvement of the skeletal system is the most common site for children and may manifests as pain, palpable mass, motion deficit, or chronic otitis in the case of mastoid or middle ear involvement. Bony lesions from LCH are predominantly lytic in appearance, and the skull is the most frequently involved bony structure.
Cutaneous involvement affects primarily the skin of the scalp and groin and resembles a seborrheic dermatitis. Patients with cranial involvement (posterior pituitary or hypothalamus) may present with diabetes insipidus (DI). The disease may also involve the cervical lymph nodes. Pulmonary involvement is more typically seen in adults. Hepatomegaly, splenomegaly, bone marrow infiltration, and involvement of the gastrointestinal tract are further potential sites of disease.
Evaluation of the patient with suspected LCH should include complete H&P. Routine laboratory work should include white cell blood count with differential. Patients who have symptoms of DI should also undergo a water restriction test. A skeletal survey with or without bone scan should be performed to assess for potential lytic lesions. Further imaging with CT of the head should be performed in patients with skull, orbital, or mastoid involvement. MRI of the head is indicated for patients with DI and those suspected of having brain parenchymal disease involvement. CT of the chest is performed to evaluate patients with pulmonary involvement. An MRI of the abdomen should be performed for patients with palpable hepatomegaly or splenomegaly.
Management Principles
Treatment of LCH depends on the site and extent of disease. Asymptomatic lesions may be observed. For patients with involvement of only the skeletal system, treatment options include curettage, excision, or intralesional steroid injection. Response rates with curettage or excision alone range from 70% to 90%.105 Single system multifocal bone disease may be effectively treated with corticosteroids or chemotherapy, such as vinblastine. For skin-only disease, topical nitrogen mustard and methotrexate are considered effective treatments.
In patients with multisystem disease with symptoms (fever, pain, failure to thrive) or organ dysfunction, treatment with systemic therapy is indicated. In LCH-II, all patients received initial treatment with prednisone and vinblastine and were randomized to intensification with or without etoposide.103,106 The patients treated with intensification demonstrated superior rapid response rates and decreased mortality rates compared with the standard arm.106 Exogenous antidiuretic hormone (ADH; vasopressin) is used to treat children with DI.
Radiotherapy
Due to the excellent response rates to nonradiotherapeutic measures, the role of RT in the treatment of LCH bony lesions has decreased. Indications for radiation therapy to bony sites include relapse after surgery, no signs of clinical healing after other interventions, pain relief, potential compromise of critical structures from an expansile lesion (i.e., cord compression), or if the bony site is not amenable to other local therapies. Collapsed vertebral lesions should not be irradiated unless they are painful. DI is another recognized potential indication for treatment with RT. When the decision is made to treat, 3D-CRT should be the technique of choice.
The target volume for patients with bony disease should encompass the abnormality seen on imaging with a small margin. For children, low doses on the order of 5 to 10 Gy given in 1.5- to 2.0-Gy daily fractions should be sufficient to control most bony lesions. Higher doses can be used in adults. In an early study by Smith et al.,107 92% of patients received total doses in the range of 4.5 to 10.0 Gy with an 87% local control rate. A study at the University of California–Los Angeles found local control rates of 88% with doses in the range of 6 to 15 Gy for previously untreated lesions and 8 to 15 Gy for recurrent lesions.108
The target volume for patients receiving treatment for DI should encompass the hypothalamus and pituitary gland. The recommended prescribed dose is 15 Gy in 1.5-Gy daily fractions. In a report from the Mayo Clinic, 36% of 28 evaluable patients responded to radiotherapy.109 The response rates were 60% (3 of 5 patients) in those treated with >15 Gy compared with 30% (7 of 23 patients) treated with doses of <15 Gy. Six patients had a complete response to therapy, five of whom received treatment within 14 days from diagnosis of DI.
Controversy still remains regarding the role of RT in the management of DI. In a retrospective series from MGH, 14 of 17 patients with DI received irradiation to the hypothalamic–pituitary axis.110 Only two of these patients had a complete response (cessation of ADH therapy), and no patients had a partial response. The others argue that treatment of DI from LCH is “no longer justified.”110
 VASCULAR DISORDERS
VASCULAR DISORDERS
Vascular disorders are broadly categorized into vascular tumors, most commonly benign hemangiomas, and vascular malformations, including arteriovenous malformations (AVMs) and cavernous hemangiomas. Radiosurgery has emerged as an important and common treatment option for AVMs. Although radiation was used commonly in the past to treat hemangiomas in children, the recognition of late effects associated with radiotherapy, especially secondary malignancies, has rendered this practice less common.
Arteriovenous Malformations
Background and Clinical Aspects
Intracranial AVMs are congenital vessel abnormalities consisting of widened arteries connected to the normal capillary bed. The nidus of an AVM is made up of tangled arteries and veins that are connected by one or more fistulas. The overall prevalence is low, affecting approximately 18 in 100,000 individuals, with age at presentation typically between 20 and 40 years old.
Clinical concern comes from the high risk of bleeding, estimated to be 2% to 4% per year. Approximately 50% of patients present with hemorrhage and 50% present with nonfocal (headache, nausea) symptoms or incidentally found focal neurologic deficits. The risk of death per bleed is up to 10%, and approximately 30% have serious morbidity associated with each bleed.
Diagnostic imaging includes angiography, which is invasive but allows for full grading of the AVM according to the Spetzler-Martin scale. MRI, MR angiography, and CT angiography are noninvasive and complementary studies that may be used to visualize the AVM.
Surgery
The goal of any therapy for AVM is to completely obliterate the nidus. Partial obliteration of the nidus does not decrease the bleeding risk. Complete surgical excision provides immediate cure but carries a risk of intraoperative bleeding, ischemic cerebrovascular accident, infection, and death. Surgery is particularly indicated for AVMs in superficial, noneloquent regions of the brain. Endovascular therapy (embolization) is not curative but may be used to decrease the risk of intraoperative bleeding or to decrease the size of the nidus before planned radiotherapy.
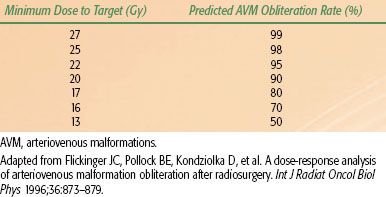
TABLE 91.6 FLICKINGER’S PREDICTED RATES OF IN-FIELD ARTERIOVENOUS MALFORMATIONS OBLITERATION BASED ON THE MINIMUM DOSE WITHIN THE TARGET VOLUME