- Incretins are gut hormones that enhance glucose-stimulated insulin secretion and have an important role in the regulation of post-prandial glucose levels.
- The major incretin hormones are glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP).
- The physiologic actions of GLP-1 include potentiation of meal-induced insulin secretion, inhibition of glucagon secretion, delay in gastric emptying and suppression of food intake and appetite.
- Beneficial effects of parenteral administration of a synthetic GLP-1 receptor agonist (exenatide or liraglutide) in patients with type 2 diabetes include improved glycemic control and mild weight loss; nausea and vomiting are the most common adverse events.
- Amylin is a naturally occurring peptide that is co-secreted with insulin from the β-cells of the pancreatic islets.
- Physiologic actions of amylin include delayed gastric emptying, suppression of post-prandial glucagon secretion and increased satiety.
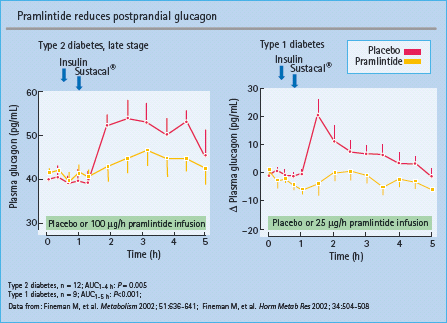
- Beneficial effects of parenteral administration of pramlintide, a synthetic amylin mimetic approved for use in insulin-treated subjects with either type 1 or type 2 diabetes, include improved glycemic control and mild weight loss; transient nausea and hypoglycemia are the most common adverse events.
- The non-insulin parenteral therapies include activators of the GLP-1 receptor, also called “incretin mimetics,“ as well as amylin mimetics.
GLP-1 receptor activators
The actions of glucagon-like peptide 1 (GLP-1) include the potentiation of meal-induced insulin secretion and, in preclinical models, proliferation and/or neogenesis of the β-cell and prevention of β-cell apoptosis has been observed. GLP-1 also inhibits glucagon secretion, delays gastric emptying and suppresses food intake and appetite. Cardiovascular effects include protection of the ischemic heart, improved left ventricular ejection fraction, endothelial function and blood pressure lowering. In animal models, GLP-1 may also possess neurotropic effects. GLP-1 receptor agonists improve glycemic control to a degree that is similar to or greater than oral antidiabetic drugs. Furthermore, they induce weight loss of 2–3 kg, on average. It is unknown whether GLP-1 receptor agonists can delay the progression of type 2 diabetes mellitus (T2DM). The GLP-1 receptor mimetics are well tolerated and the most common adverse events are gastrointestinal, including nausea and vomiting. The available GLP-1 receptor agonists require injection once or twice daily, but once-weekly GLP-1 receptor activators are in development.
The incretin effect
“The incretin effect” designates the amplification of insulin secretion elicited by hormones secreted from the gastrointestinal tract. It is quantified by comparing insulin responses to oral and intravenous glucose administration, where the intravenous infusion is adjusted so as to result in the same (isoglycemic) peripheral plasma glucose concentrations [1,2]. In healthy subjects, the oral administration of gluocose causes a two- to threefold larger insulin response than the intravenous route as a result of the actions of the “incretin hormones.” Because of this, the incretin hormones are thought to have a particularly important role in the regulation of post-prandial glucose levels.
The two most important incretins are glucose-dependent insulinotropic polypeptide (GIP), previously designated gastric inhibitory polypeptide, and GLP-1. Both have been established as important incretin hormones in mimicry experiments in humans, where the hormones were infused together with intravenous glucose to concentrations approximately corresponding to those observed during oral glucose tolerance tests. Both hormones powerfully enhanced insulin secretion, to an extent that could fully explain the insulin response [3,4], and recent experiments have documented that both are active with respect to enhancing insulin secretion from the beginning of a meal (even at fasting glucose levels), and that they contribute almost equally, but with the effect of GLP-1 predominating at higher glucose levels [5]. The effects of the two hormones with respect to insulin secretion have been shown to be additive in humans [6].
The incretin effect in T2DM
Original studies by Nauck et al. [7] and several subsequent studies have indicated that the incretin effect is severely reduced or lost in patients with T2DM [8,9]. Thus, there is little doubt that a defective incretin effect contributes to the glucose intolerance of these patients.
Given that GLP-1 and GIP are the most important incretin hormones, it is possible to analyze their contribution to the defective incretin action in patients with diabetes. Such contributions could consist of defects with respect to secretion, action or metabolism. Detailed studies of the secretion of GIP and GLP-1 in response to mixed meals in patients with T2DM revealed a slightly impaired secretion of GIP, but a more pronounced impairment with respect to the secretion of GLP-1. The reduction was particularly prominent during the second and third hour of the meal test, while the initial response was unimpaired. The decreased response was related to both body mass index (BMI; the higher the BMI, the lower the incretin response) and to the actual diabetic state [10]. A decreased GLP-1 secretion in obese subjects has been observed repeatedly [11] and is related to an impaired incretin effect [12,13]. Insulin resistance is also independently related to a decreased GLP-1 meal response [13,14]. A decreased secretion of GLP-1 may not be seen in all obese or insulin-resistant subjects. Experience from the authors’ laboratory suggests that in addition to insulin resistance and obesity, longer duration of diabetes and, in particular, influence of therapy is important. For instance, it is now clear that metformin enhances GLP-1 secretion [15]. When present, impaired secretion of GLP-1 is likely to contribute to the failing incretin effect.
The metabolism of GLP-1 and GIP was compared by Vilsb0ll et al. [16,17] in patients with diabetes and controls; both hormones were metabolized at similar rates, suggesting that differences in their elimination do not explain the reduced plasma concentrations in diabetes.
With respect to the actions of the incretin hormones, it was discovered in 1993 that infusion of large amounts of GLP-1 resulted in near-normal insulin responses in patients with T2DM, whereas GIP had no significant effect [18]. However, infusions of GIP and GLP-1 at rates resulting in physiologic elevations of the incretin levels (as observed after mixed meal ingestion) while glucose was clamped at 15 mmol/L, failed to affect insulin secretion at all, despite these infusion rates greatly (and equally) increasing insulin secretion in healthy subjects [19]. These findings illustrate the dramatic loss of potency of both GIP and GLP-1 in T2DM. By contrast, when infused to supraphysiologic levels, GLP-1, but not GIP, was capable of restoring the β-cell sensitivity to glucose to completely normal values [20]. The mechanisms whereby GLP-1, but not GIP, stimulates insulin secretion in T2DM remain unknown, but the observation suggested that GLP-1 in pharmacologic doses could be used clinically to enhance insulin secretion in T2DM.
Finally, one may ask whether the incretin defect is a primary event, perhaps a major etiologic contributor to the β-cell failure that characterizes T2DM. Several observations suggest that this is not the case [9], but may be related to insulin resistance and/or hyperglycemia [21], as reviewed recently [9]. Interestingly, there are now data to suggest that intensified treatment resulting in near-normal glucose levels may lead to a partial restoration of incretin action of GLP-1 and GIP [19,22] and induction of insulin resistance in healthy subjects is associated with a loss of the incretin effect [23].
Actions of GLP-1
The acute insulinotropic effects of GLP-1 raised interest in the use of this peptide for the treatment of diabetes. In addition, the peptide possesses a number of other effects, which in the context of diabetes treatment must be considered favorable (Figure 30.1).
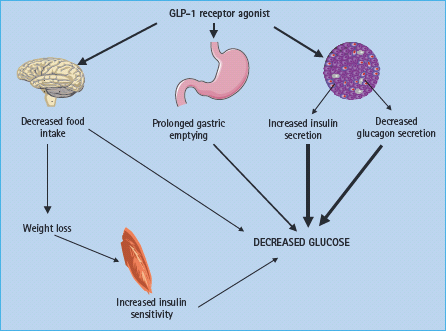
Effects on the islets
The insulinotropic activity of GLP-1, which represents a potentiation of glucose-induced insulin secretion and therefore is strictly glucose-dependent, is, at least partly (see below), exerted via interaction with the GLP-1 receptor located on the cell membrane of the β-cells [24]. Binding of GLP-1 to the receptor causes activation of adenylate cyclase resulting in the formation of cyclic AMP (cAMP), via a stimulatory G-protein. Most of the actions of GLP-1 are secondary to the formation of cAMP and results in a plethora of events including altered ion channel activity, intracellular calcium handling and enhanced exocytosis of insulin-containing granules [25]. The effects of glucose and GLP-1 may converge at the level of the KATP channels of the β-cells. These channels are sensitive to the intracellular ATP levels and, thereby, to glucose metabolism of the β-cells, but may also be closed by protein kinase A activated by GLP-1, resulting in subsequent depolarization of the plasma membrane and opening of voltage-sensitive calcium channels [26–28]. This has clinical importance because sulfonylurea drugs, which bind to and close the KATP channels and thereby cause membrane depolarization and calcium influx, may uncouple the glucose dependency of GLP-1 [29]. Indeed, 30–40% of patients treated with both sulfonylurea compounds and a GLP-1 agonist (see below) experience, usually mild, hypoglycemia.
The transcription factor PDX-1, a key regulator of islet growth and insulin gene transcription, appears to be essential for most of the glucoregulatory, proliferative and cytoprotective actions of GLP-1 [30]. In addition, GLP-1 upregulates the genes for the cellular machinery involved in insulin secretion, such as the glucokinase and GLUT-2 genes [31]. For further discussion of the insulinotropic effects of GLP-1, see Holst [32].
Importantly, GLP-1 also has trophic effects on β-cells [33]. Not only does it stimulate β-cell proliferation [34,35], but it also enhances the differentiation of new β-cells from progenitor cells in the pancreatic duct epithelium [30,36] and inhibits apoptosis of β-cells including human β-cells [37,38]. Because the normal number of β-cells is maintained in a balance between apoptosis and proliferation [39], this observation is of considerable interest, and also raises the possibility that GLP-1 could be useful in conditions with increased β-cell apoptosis.
The complicated mechanisms whereby GLP-1 may exert trophic effects on the β-cells were reviewed recently by Drucker [40].
Reflex activation of the β-cell
GLP-1 is extensively degraded by the enzyme dipeptidyl peptidase 4 (DPP-4) and it has been demonstrated that only about one-quarter of the amount of GLP-1 leaving the gut (the endocrine organ) is still in the intact active form, and only about 8% of what is secreted actually reaches the pancreas as the intact peptide [41–43]. This has given rise to speculation that the actions of GLP-1 on the β-cells might be exerted indirectly via activation of long vagovagal reflexes [42]. Indeed, the GLP-1 receptor is expressed in cell bodies in the nodose ganglion from which the sensory afferents from the gastrointestinal tract emanate [44], and it has been demonstrated that blockade of the autonomic ganglia also blocked the effects of intraportally injected GLP-1 on insulin secretion [45]. Thus, it may be that under physiologic conditions the majority of the effects of GLP-1 on insulin secretion are transmitted via autonomic nerves.
Effects on glucagon secretion
GLP-1 also strongly inhibits glucagon secretion. Because in patients with T2DM there is fasting hyperglucagonemia as well as exaggerated glucagon responses to meal ingestion [46], and because it has been demonstrated that the hyperglucagonemia contributes to the hyperglycemia of patients [47], this effect may be as important as the insulinotropic effects. The mechanism whereby GLP-1 inhibits glucagon secretion remains unsettled but there is evidence that the somatostatin-producing D-cells of the islets transmit the effects of GLP-1 by paracrine inhibition of the a-cells [48].
Effects on the gastrointestinal tract
Further important effects of GLP-1 include inhibition of gastrointestinal secretion and motility, notably gastric emptying [49,50]. By this mechanism, GLP-1 may curtail post-prandial glucose excursions [51]. and thereby reduce the number of episodes with high post-prandial glucose levels. There has been concern that the powerful inhibitory effect could represent a problem in patients with gastroparesis, but so far there have been no reported cases.
Effects on appetite and food intake
GLP-1 inhibits appetite and food intake in normal subjects [52] as well as in obese subjects with T2DM [53,54], and it is thought that GLP-1 is one of the gastrointestinal hormones that normally regulate food intake [32]. Clinical studies have shown that the effects on food intake are maintained for several years and lead to a sustained or progressive weight loss.
Cardiovascular effects
It has been known for some time that there are GLP-1 receptors in the heart [55]. A physiologic function for these receptors was indicated in recent studies in mice lacking the GLP-1 receptor, which exhibit impaired left ventricular contractility and diastolic functions as well as impaired responses to exogenous epinephrine [56]. GLP-1 also increases left ventricular pressure and coronary blood flow in isolated mouse hearts [57], although in normal rat hearts, it reduces contractility [58]. However, further studies in rats showed that GLP-1 protects the ischemic and reperfused myocardium in rats by mechanisms independent of insulin [58,59]. Protective effects may be demonstrated by administration of GLP-1or GLP-1 receptor agonists both before and after ischemia (Figure 30.2) [57,60], and may involve the p70s6 kinase [61]. Surprisingly, some of the cardiac effects may also be elicited by the metabolite GLP-1 9–36 amide, which is formed rapidly in the circulation, but which has a strongly reduced activity on the classic GLP-1 receptor of the β-cells [57,60,62]. It has therefore been suggested that a different receptor mediates at least some of the cardiovascular effects, although not all studies support this [61]. Whatever the mechanism, these findings may have important clinical implications. Nikolaidis et al. [63] studied patients treated with angioplasty after acute myocardial infarction, but with postoperative left ventricular ejection fractions as low as 29%. In these patients, GLP-1 administration significantly improved the ejection fraction to 39% and improved both global and regional wall motion indices [63]. In studies of dogs with induced dilated cardiomyopathy, GLP-1 was reported to improve left ventricular and systemic hemodynamics dramatically, and it was suggested that GLP-1 may be a useful metabolic adjuvant in decompensated heart failure [64]. Indeed, addition of GLP-1 to standard therapy in patients with heart failure significantly improved left ventricular ejection fraction, myocardial oxygen uptake, 6-minute walking distance and quality of life [65]. In another study in which patients undergoing coronary artery bypass grafting were randomized to receive GLP-1 or conventional treatment, there was less use of inotropic and vasoactive drugs and fewer arrhythmias, as well as better glycemic control, in the GLP-1 group [66]. The cardioprotective actions of GLP-1 were recently reviewed by Fields et al. [67].
Figure 30.2 Cardioprotective effect of the GLP-1 receptor agonist, exendin-4. Post-conditioning with the GLP-1 receptor agonist, exendin-4 (i.e. addition of the agonist to the perfusate after the ischemia period) reduces infarct size in an isolated rat heart model of ischemia-reperfusion injury. Adapted from Sonne et al. [60]
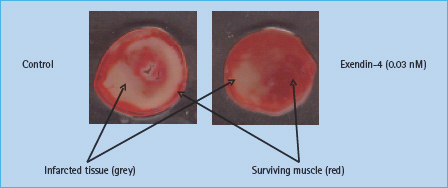
Furthermore, GLP-1 has been found to improve endothelial dysfunction in patients with T2DM and coronary heart disease, again a finding with interesting therapeutic perspectives [68]. An effect on endothelial dependent vasodilation was confirmed in healthy subjects [69], and functional receptors for GLP-1 have been identified on endothelial cells [57]. Chronic administration of GLP-1 agonists for clinical use is generally associated with small but significant decreases in blood pressure [70,71]. This may reflect the vasodilatory actions of GLP-1 as observed in various animal experimental models [57,72].
Neurotropic effects
Intracerebroventricular GLP-1 administration was associated with improved learning in rats and also displayed neuroprotective effects [73,74]. GLP-1 has also been proposed as a new therapeutic agent for neurodegenerative diseases, including Alzheimer disease [75]. supported by observations that GLP-1 can modify processing of amyloid precursor protein and protect against oxidative injury [76]. Very recently, it was reported that the GLP-1 receptor agonist, exendin 4, promoted adult neurogenesis, normalized dopamine imbalance and increased the number of dopaminergic neurons in the substantia nigra in animal models of Parkinson disease [77].
Actions of GLP-1 in T2DM
In agreement with the findings of preserved insulinotropic actions of GLP-1 in T2DM [18], intravenous infusion of GLP-1 at 1.2 pmol/kg/minute was demonstrated to be capable of completely normalizing plasma glucose in patients with long-standing severe disease admitted to hospital for insulin treatment [78]. Subsequent studies in patients with moderate disease showed that plasma glucose concentrations could be near-normalized by an intravenous GLP-1 infusion covering the night-time and the next day, including two meals [79]. Continuous intravenous infusion of GLP-1 is not clinically practical and the effect of subcutaneous injections of GLP-1, given to both patients and healthy subjects [80], on plasma glucose and insulin concentrations turned out to be very short-lasting, because of an extremely rapid and extensive metabolism of GLP-1 in the body [81,82]. The intact peptide has an apparent intravenous half-life of 1–2 minutes and a plasma clearance amounting to 2–3 times the cardiac plasma output [16]. GLP-1 degradation is brought about by the actions of the ubiquitous enzyme, DPP-4, which catalyzes the removal of the two N-terminal amino acids of the molecule, thereby rendering it inactive [81] with respect to insulin secretion (but it may have cardiovascular effects, see above). The metabolic instability of GLP-1 clearly restricts its clinical usefulness, but Zander et al. [83] carried out a clinical study in which GLP-1 or saline was administered as a continuous subcutaneous infusion (using insulin pumps) for 6 weeks to a group of patients with T2DM. The patients were evaluated before, after 1 week and after 6 weeks of treatment. No changes were observed in the saline-treated control group, whereas in the GLP-1 group, fasting and average plasma glucose concentrations were lowered by approximately 4–6 mmol/L, HbA1c decreased by 1.2% (13 mmol/mol), free fatty acids were significantly lowered and the patients had a gradual weight loss of approximately 2 kg. In addition, insulin sensitivity, determined by a hyperinsulinemic euglycemic clamp, almost doubled, and insulin secretion capacity (measured using a 30mmol/L glucose clamp + arginine) greatly improved. There was no significant difference between results obtained after treatment for 1 or 6 weeks, but there was a tendency towards further improvement of plasma glucose as well as insulin secretion. There were very few side effects and no differences between saline and GLP-1 treated patients in this respect. This study therefore provided “proof of concept” for the principle of GLP-based therapy of T2DM, and further attempts to utilize the therapeutic potential of GLP-1 have included, on one hand, the development of stable, DPP-4 resistant analogs [84] and, on the other hand, inhibitors of DPP-4 demonstrated to be capable of protecting the peptide from degradation and thereby augmenting its insulinotropic activity (see Chapter 29) [85].
GLP-1 receptor agonists
None of the per oral antidiabetics or insulin have been shown to stop the progressive decline of β-cell function, and none address the elevated glucagon secretion [86]. Some oral antidiabetics are associated with weight gain (sulfonylureas, glinides and glitazones), hypoglycemia (sulfonylureas and glinides), lactate acidosis (metformin), intestinal side effects (metformin and a -glucosidase inhibitors) or peripheral edema and fractures (glitazones) [86].
In contrast, GLP-1 receptor agonists regulate glucose metabolism and promote weight loss with a low risk of hypoglycemia. At present two GLP-1 receptor agonists are approved. Exenatide (Byetta®) was approved by the US Food and Drug Administration (FDA) in April 2005 and released in Europe in 2007 for the treatment of T2DM. Liraglutide (Victoza®), a once-daily human GLP-1 analog, was approved in Europe in May 2009 and by the FDA in January 2010.
Exenatide (Byetta)
Exendin-4, originally isolated from the saliva of the Gila monster, has 53% sequence homology with human GLP-1 in its first 30 amino acids [87]. Synthetic exendin-4, referred to as exenatide (1–39), is resistant to DPP-4 cleavage, and has a subcutaneous half-life in humans of approximately 2.5 hours after injection and a peak action 2–3 hours after injection [88], as reviewed in [89]. Exenatide is not recommended for patients with severe kidney failure (creatinine clearance <30mL/minute), because it is predominantly eliminated by glomerular filtration with subsequent proteolytic degradation. Exenatide, administered twice daily within 60 minutes of morning and evening meals, lowers glucose for 5–7 hours and predominately affects post-prandial glycemic excursion, with only modest effects on lunch hyperglycemia and on fasting blood glucose [89–90]. The main side effect, nausea, is dose-dependent after subcutaneous injection, but may show some tachyphylaxis [88]. Therefore, it is recommended to start therapy with 5 |ig twice daily for 1 month and subsequently to increase to 10 |ig twice daily.
In clinical studies, exenatide exhibited actions that are similar to those of GLP-1, but the in vivo glucoregulatory potency of exenatide is significantly greater than that of native GLP-1 [91]. Increased insulin secretion, inhibition of glucagon secretion and delayed gastric emptying are the main mechanisms by which exenatide improves glucose metabolism (Figure 30.1) [88,89,92].
Clinical efficacy of exenatide
In the three so-called AMIGO (Diabetes Management for Improving Glucose Outcome) trials, the efficacy of exenatide in combination with metformin, sulfonylurea or a combination of the two was studied for 30 weeks [93–95]. Basal mean HbA1c in the three studies was 8.3–8.6% (67–70mmol/mol). The placebo-corrected reduction in HbA1c with 10|ig b.i.d. was 0.9–1.0% (10–11 mmol/mol), and the weight loss was 1.6–2.6kg after 30 weeks [94–95]. Body weight reductions were directly proportional to baseline BMI, with about 80% of subjects losing weight [93–95]. Improvements were observed in some cardiovascular risk factors and hepatic biomarkers [71]. Thus, increases in high density lipoprotein (HDL) cholesterol (+0.12 mmol/L), decreases in triglycerides (–0.43 mmol/L) and diastolic blood pressure (–2.7mmHg) were achieved [71]. Two weeks’ treatment with exenatide was associated with significantly decreased post-prandial triglycerides excursions [96].
In patients with T2DM treated with a glitazone and metformin, exenatide reduced HbA1c (baseline mean HbA1c 7.9%, 63 mmol/ mol) by 1.0% (11 mmol/mol) and weight by 1.6kg compared with placebo [97]. No clinically significant episodes of hypoglycemia were reported [97]. As in most of the other studies, the drop-out rate in the exenatide group was twice that in the placebo group (29% vs 14%) [97].
In an open-label trial, insulin glargine once daily or exenatide twice daily were added for patients inadequately controlled by metformin and sulfonylurea [98]. HbA1c fell by 1.1% (12 mmol/ mol) points in both groups (baseline mean HbA1c 8.2%, 66 mmol/ mol) [98]. Body weight increased by 1.8 kg with insulin glargine while a 2.3-kg weight loss was obtained with exenatide. Frequencies of hypoglycemia did not differ between treatments, but nocturnal hypoglycemia was less frequent with exenatide. Approximately 19% and 10% of the patients withdrew from the study in the exenatide and glargine groups, respectively [98]. In a 52-week trial, exenatide was compared with twice daily biphasic insulin aspart (baseline mean HbA.c 8.6%, 70mmol/mol) [99]. A greater proportion of exenatide-treated patients reached HbA1c <7.0% (<53mmol/mol, 32% vs 24%) [99]. Bodyweight changes (-2.5 kg vs +2.9 kg) also favored exenatide over insulin. Systolic (–5 mmHg) and diastolic (–2 mm Hg) blood pressures were significantly reduced with exenatide, but remained unchanged with insulin. No difference with respect to hypoglycemia was found between the two groups [99].
In obese subjects with T2DM treated with insulin plus oral antidiabetic drugs, addition of exenatide induced a 6.5-kg weight loss and lowered HbA1c by 0.6% (7 mmol/mol) points over a 26-week follow-up period [100]. Rapid acting and premix insulin doses were reduced by 28% and 60%, respectively, while the dose of basal insulin was unchanged. In contrast, substitution of exenatide for insulin in insulin-treated patients with T2DM appears to be less successful, because such a switch resulted in a slight increase in HbA1c (+0.3%, 3mmol/mol) [101].
Other studies have shown that exenatide improved first-phase insulin responses to intravenous glucose and decreased the proinsulin: insulin ratio, indicating a beneficial effect of exenatide treatment on β-cell function [94,102]. Nevertheless, no human data indicate that exenatide can protect or restore the β-cell mass. Thus, the β-cell function did not change from before start of treatment when tested after 1 year treatment with exenatide followed by 1 month wash-out of exenatide [103].
Adverse effects with exenatide
Exenatide has generally been well tolerated and rarely causes treatment discontinuation because of side effects, but in the phase 3 studies drop-out rates in the exenatide groups were twice those in the placebo-treated groups [93–95]. Hypoglycemia is not a problem when exenatide is used in monotherapy or in combination with metformin [94], but mild hypoglycemia was observed in 15–36% of subjects receiving sulfonylurea in combination with exenatide [93,95]. Appropriate glucagon responses have been demonstrated during treatment with GLP-1 or exenatide, which may protect against severe hypoglycemia [104,105]. Mild to moderate nausea has been reported in up to 57% of patients treated with exenatide compared to about 7–23% on placebo in combination with oral antidiabetic drugs [93–95]. Nausea generally occurs during the first days of treatment and, in most patients, decreases with time. Vomiting and headache have also been observed in some patients. Other adverse effects reported include diarrhoea, dizziness and constipation [106]. About 40–50% of treated subjects develop antibodies to exenatide [93–95]; the importance of these is unknown, but they do not seem to influence efficacy on glucose control, except in some subjects with very high titers. Exenatide treatment has been associated with acute pancreatitis, and clinicians should be alert to this potential adverse effect [107]; however, it is not evident that the incidence of acute pancreatitis is higher in those receiving exenatide than in the background diabetic population. It has been recommended that exenatide should not be used in subjects at risk of pancreatitis (e.g. with alcoholism, cholecystolithiasis or hypertriglyceridemia), and if pancreatitis is suspected the drug should be discontinued.
Liraglutide
Liraglutide is a once daily human GLP-1 analog, with a half-life of about 13 hours following subcutaneous injection. Liraglutide is based on the structure of native GLP-1 with an amino acid substitution (lysine with arginine at position 34) and attachment of a C16 acyl chain via a glutamoyl spacer to lysine at position 26 [108]. Liraglutide is relatively stable towards DPP-4 degradation both intrinsically (because it may form heptameric aggregates) and as a consequence of binding to albumin [108]. After initiation of treatment, steady-state concentrations are obtained after 3–4 days [108].
Studies with liraglutide
In the clinical Liraglutide Effect and Action in Diabetes (LEAD) development program the initial dose of liraglutide was escalated weekly in a stepwise fashion from 0.6 mg/day to 1.2 mg/day, and in some patients to 1.8 mg/day. In monotherapy (basal mean HbA1c 8.3%, 67mmol/mol) liraglutide 1.2 and 1.8mg reduced HbA1c more than glimepiride (LEAD-3) over 52 weeks (0.84%, 1.14% [12 mmol/mol] and 0.5% [5 mmol/mol], respectively) [109]. The decrease in HbA1c with 1.8 mg liraglutide was significantly greater than with 1.2 mg liraglutide, and the decrease was most pronounced in the group previously treated with only lifestyle changes (1.2mg liraglutide, 1.2% [13mmol/mol]; 1.8mg liraglutide, 1.6% [17 mmol/mol]; and glimepiride, 0.9% [10 mmol/mol]) [109]. A weight decrease occurred in the liraglutide groups, compared to weight gain in the glimepiride group, resulting in a weight difference of 3.2–3.6 kg [109]. Nausea, mostly transient, occurred in 29% of the participants in the liraglutide groups compared with 9% in the glimepiride group. The rates of minor hypoglycemic episodes were significantly lower for the liraglutide groups versus glimepiride (12, 8 and 24 events/subjects/year) [109]. Two-year data showed that liraglutide had a long-lasting beneficial effect on glycemic control with a reduction in HbA1c of –0.9% (10mmol/mol), –1.1% (12mmol/ mol) and –0.6% (7mmol/mol) in the 1.2mg liraglutide, 1.8mg liraglutide and glimepiride groups, respectively [110]. The weight loss was –2.1kg, –2.7kg and +1.1kg in the three groups [110].
Liraglutide, when added to glimepiride (LEAD-1) reduced HbA1c more than rosiglitazone over 26 weeks (1.1% vs 0.4%, 12 vs 4 mmol/mol) [111]. The changes in weight were superior for liraglutide compared with rosiglitazone [111].
Liraglutide was compared with 4 mg glimepiride when added to metformin (baseline mean HbA1c 8.4%, 68 mmol/mol) in LEAD-2 [112]. The reduction in HbA1c did not differ between glimepiride and liraglutide over 26 weeks (1.0% vs 1.0%, 11 mmol/ mol). The reduction in weight in the two groups treated with 1.2 or 1.8 mg/day liraglutide was 2.6 and 2.8 kg, compared with an increase in weight of 1.0 kg in group the treated with glimepiride [112]. Mild hypoglycemic events were reported in up to 2.5% of the patients treated with metformin and liraglutide compared with 17% in the glimepiride group [112]. In LEAD-4, liraglutide, when added to metformin and rosiglitazone, reduced HbA1c (–1.5% vs +0.5%, –16 vs +5 mmol/mol) and weight (–2.0 vs +0.6 kg) more than placebo [113].
In LEAD-5, patients treated with metformin plus a sulfonylurea were randomized to treatment with liraglutide (1.8mg) or insulin glargine [114]. Mean reduction in HbA1c was significantly greater with liraglutide than in insulin glargine treated groups (1.1% vs 0.9%, 12 vs 10mmol/mol), and more than half of the patients treated with liraglutide reached the American Diabetes Association (ADA) target of HbA1c <7.0% (<53 mmol/mol), and 36% reached HbA1c <6.5% (<48 mmol/mol) [114]. With respect to body weight, liraglutide resulted in a weight difference of 3.5 kg compared to the insulin glargine treated group [114]. Five patients (2.2%) experienced major hypoglycemic episodes in the liraglutide treated group; there were no major episodes in the insulin glargine treatment group. It is well described that the combination of a sulfonylurea with a GLP-1 receptor agonist increases the risk of hypoglycemia. The mechanism behind this reflects an uncoupling of the glucose-dependent insulin secretion of GLP-1 when combined with a sulfonylurea [29]. Incidence of minor hypoglycemic events did not differ between the groups [114].
In LEAD-6, a head-to-head comparison of 1.8 mg liraglutide once daily with exenatide 10 μg twice daily, the reduction in HbA1c was 1.1% (12mmol/mol), compared to 0.8% (9mmol/ mol), respectively [115]. Both groups lost around 3 kg in weight. The most frequent reported adverse events for both liraglutide and exenatide treated groups were nausea at a level of around 25% when initiating therapy [115]. After week 8–10 the percentage of patients reporting nausea with liraglutide was well below 10%, while in the exenatide group the level remained at about 10%. The rate of minor hypoglycemia was significantly lower in the liraglutide group. After 30 weeks the patients treatment with exenatide were shifted to liraglutide, which resulted in a further 0.3% (3 mmol/mol) reduction in HbA1c.
Treatment with liraglutide has been associated with improvement of several cardiovascular risk factors: C-reactive protein (CRP), tumor necrosis factor a (TNF-α) induced plasminogen activator inhibitor 1 (PAI-1), vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1) and B-type natriuretic peptide (BNP) [116]. The weight reduction in the clinical studies depend on basal BMI, and was in subjects with BMI <25kg/m2 from 0–2 kg increasing to 1–4.5 kg in subjects with a BMI >35 kg/m2 [117]. The greatest weightloss was observed with the combination of liraglutide and metformin, which may reflect that metformin is weight neutral while sulfonylureas such as glimepiride and rosiglitazone are associated with weight gain [117]. The highest dose of liraglutide (1.8 mg/day) reduces systolic blood pressure by 2.7–4.5 mmHg versus comparator treatment, but in patients with a systolic blood pressure greater than 140 mmHg the reduction was about 11 mmHg [118]. Lastly, liraglutide improved β-cell function evaluated by homeostasis model assessment (HOMA) and the proinsulin: insulin ratio [119]. The thyroid C-cell neoplastic changes, which have been observed in rodents and mice, have not been observed in humans [120]. Five cases of pancreatitis were described during the phase 3 program with liraglutide.
Comparison of GLP-1 receptor agonists with DPP-4 inhibitors
The GLP-1 receptor agonist and DPP-4 inhibitors differ in route of administration and may have differing side effect and tolerability profiles (Figure 30.3). Two head-to-head studies have compared the efficacy of a GLP-1 receptor agonist and the DPP-4 inhibitor sitagliptin. Liraglutide reduced HbA1c by 1.4% vs 0.9% (14 vs 10mmol/mol) and weight by –3.5 vs –0.9kg compared with sitagliptin. In another study, exenatide once weekly reduced HbA1c by 1.7% vs 1.0% (19 vs 11 mmol/mol), and the weight by 2.8 vs 0.9 kg compared with sitagliptin [121].
Figure 30.3 Dose-response relationships for the effects of GLP-1. The figure illustrates the relationship between the plasma concentrations of GLP-1 or GLP-1 mimetics and their clinical effects and side effects as observed during treatment with GLP-1 mimetics or incretin enhancers (DPP-4 inhibitors, see Chapter 29). With modestly elevated GLP-1 levels, as obtained with both mimetics and enhancers, there are significant effects on the pancreatic islets. Higher concentrations (which may not be reached during therapy with enhancers) are needed to slow down gastric emptying and to reduce appetite and food intake. At even higher concentrations, which may be reached with GLP-1 mimetics, side effects such as nausea, diarrhea and vomiting might result. Adapted from Holst JJ, Deacon CF, Vilsboll T, Krarup T, Madsbad S: Glucagon-l ike peptide-1, glucose homeostasis and diabetes. Trends Mol Med 2008; 14: 161–168.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


