An increased risk of NHL has been associated with a number of environmental exposures and/or disease states (see Table 103.1).12,13 There is controversial evidence that certain chemical exposures, specifically the herbicide phenoxyacetic acid, increase the risk of NHL.14 Other potential environmental associations include exposure to arsenic, pesticides, fungicides, chlorophenols, or organic solvents, halomethane, lead, vinyl chloride, or asbestos.15,16 Occupational exposures associated with an increased risk include agricultural work, welding, and work in the lumber industry.17 NHL has been observed as a late complication of prior chemotherapy and/or radiation therapy. Specifically, patients with HL treated with radiation therapy and chemotherapy exhibit an increased risk of developing secondary DLBCL.18
Diseases of inherited and acquired immunodeficiency as well as autoimmune diseases are associated with an increased incidence of lymphoma (see Table 103.1). The association between immunosuppression and induction of NHLs is compelling because, if the immunosuppression can be reversed, a percentage of these lymphomas regress spontaneously.19 The incidence of NHL is nearly 100-fold increased for patients undergoing organ transplantation necessitating chronic immunosuppression, and is greatest in the first year posttransplant. About 30% of these arise as a polyclonal B-cell proliferation that evolves into a clonal B-cell malignancy. The NHLs that occur in the context of immunosuppression or immunodeficiency, including human immunodeficiency virus 1 (HIV-1) infection, are frequently associated with EBV.20 Histologically, DLBCLs are most frequently associated with immunosuppression and autoimmune diseases, although almost all histologies can be seen. The rare inherited immunodeficiency diseases (X-linked lymphoproliferative syndrome, Wiskott-Aldrich syndrome, Chédiak-Higashi syndrome, ataxia telangiectasia, and common variable immunodeficiency syndrome) are complicated by highly aggressive lymphomas. The elevated incidence of lymphoma in iatrogenic immunosuppression, AIDS, and autoimmune disease argues strongly for immune dysregulation contributing in the pathogenesis of some lymphomas.21,22 An increased risk of NHL has been observed in first-degree relatives with NHL, HL, or chronic lymphocytic leukemia (CLL). In large databases studies, about 9% of patients with lymphoma or CLL have a first-degree relative with a lymphoproliferative disorder.23,24
BIOLOGIC BACKGROUND FOR CLASSIFICATION OF LYMPHOID NEOPLASMS
Current lymphoma classification systems divide the lymphomas into different entities based, in part, on their perceived cell of origin (Fig. 103.1, Table 103.2). During embryogenesis, hematopoietic stem cells (HSC) from the liver and the placenta give rise to progenitor cells that migrate to the thymus and bone marrow where they undergo a program of antigen-independent differentiation into T- and B-cell lineage precursor cells, directed largely by the microenvironment.25 Postnatally, all lymphoid cells are derived from bone marrow HSCs, which give rise to very early lymphoid progenitors with B, T, and NK lymphocyte potential. These cells, in turn, yield B-cell progenitors in the marrow, the site of early stages of B-cell differentiation, as well as other progenitors that migrate to the thymus and undergo T-cell differentiation.
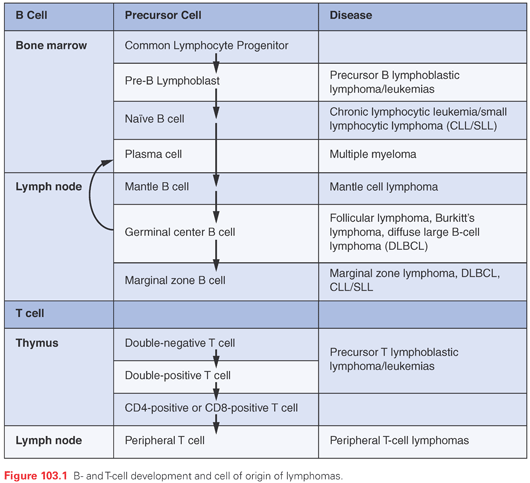
B-Cell Development
The initial commitment to B-cell differentiation by lymphoid progenitors in the bone marrow requires the expression of the master B lineage transcription factor PAX5, which directly upregulates the expression of early B lineage markers such as CD19.26 Subsequent precursor B-cell development depends on a transcriptional program that is driven by PAX5 and downstream transcription factors and prosurvival signals produced by stepwise rearrangement of immunoglobulin (Ig) genes, which requires the lymphoid-specific recombination factors RAG1 and RAG2 and also involves the specialized DNA polymerase terminal deoxynucleotidyl transferase (TdT).27 During development, pre-B cells pass through checkpoints that correspond to specific stages of Ig gene assembly, beginning with rearrangement of the Ig heavy chain locus (IgH).28 Productive (in-frame) rearrangement of IgH leads to the expression of IgM heavy chain, which combines with a lambdalike polypeptide to enable the assembly of pre–B-cell receptors. The pre–B-cell receptor generates signals that prevent apoptosis, turns off further IgH gene rearrangement (contributing to allelic exclusion, the expression of only a single IgH in each B-cell clone), and turns on rearrangements of the Ig light chain loci, first the kappa loci and, if these rearrangements are nonproductive, then the lambda light chain loci. During the period of Ig gene rearrangement, pre-B cells lack complete surface Ig and express CD19 and CD10, the common acute lymphoblastic leukemia antigen.29 Precursor B cells that productively rearrange one or another light chain locus express the surface Ig receptor (sometimes referred to as the B-cell receptor [BCR]), which also transmits key survival signals that prevent apoptosis. Cells that express surface Ig upregulate additional B-cell markers such as CD79a, cytoplasmic and surface CD22, and CD20, as well as prosurvival factors such as BCL2, and downregulate CD10 and TdT emerging from the process as mature, immunologically naïve B cells.
In mice, two major types of naïve B cells have been defined. Roughly 90% of circulating and tissue-based B cells fall into the B2 class. B2 cells are widely distributed and largely respond to antigens in a T-cell–dependent fashion, a process that yields class-switched plasma cells expressing high-affinity Ig. B1 cells can be further subdivided into those that do or do not express the antigen CD5.30 CD5+ B1a cells produce broadly reactive natural IgM, whereas CD5− B1b cells can generate T-independent, long-lasting memory-type IgM responses to some infectious pathogens. Whether B1 cells exist as a distinct B-cell lineage in humans has been (and remains) controversial, but it is notable that some human B-cell tumors, particularly CLL, is comprised of cells bearing some similarity to murine B1 cells.
From the marrow, naïve B cells migrate through the blood and extravasate into secondary lymphoid tissues, such as the spleen, the lymph nodes, and mucosa-associated lymphoid tissues in the gut. Homing of B cells to specific tissues appears to be controlled largely by chemokines that activate chemokine receptors expressed on B cells.31 Upon encountering an antigen in peripheral tissues, B cells may either be induced to differentiate directly into short-lived IgM secreting plasma cells or may migrate to B-cell follicles. Antigen-mediated B-cell activation requires the transcription factor MYC and is accompanied by an increase in cell size and entry into cell cycle.32,33 Once in follicles, the B cells downregulate MYC and BCL2 and upregulate the transcriptional repressor BCL6, which, like MYC, is essential for secondary B-cell follicle formation; secondary follicles are also known as germinal centers.34 Downregulation of BCL2 may permit the elimination of B cells making low affinity antibodies, and in fact, most B cells entering into the germinal centers undergo apoptosis and are phagocytosed by resident macrophages (often referred to as tingible body macrophages because they contain a readily visible nuclear fragment derived from defunct B cells). The key roles of MYC, BCL2, and BCL6 in this process explain why the genes encoding these factors are commonly mutated in B-cell lymphomas (discussed later). Follicular B cells also upregulate the expression of activation-induced cytosine deaminase (AID), a gene product required for both somatic hypermutation and Ig class switching. Cells that by chance acquire mutations that increase Ig affinity for an antigen survive thanks to signals transmitted through the Ig receptor and go on to undergo class-switching, a process that is regulated by cytokines.35 The germinal center reaction also requires follicular dendritic cells and a special class of CD4-positive follicular T cells that express the CD40 ligand.36 B cells that survive this process may leave the germinal centers to take up residence in surrounding marginal zones to become long-lived memory B cells, or may terminally differentiate into plasma cells, which may take up residence in the medulla of the lymph nodes or the red pulp of the spleen, or home back to the bone marrow.
It is notable that the most common human lymphomas are B-cell tumors composed of lymphocytes with somatically mutated Ig genes, an alteration that marks these tumors as having arisen from cells that have experienced a germinal center reaction. Many of these same tumors also have mutations that bear the molecular hallmarks of mistakes that occurred during attempted somatic hypermutation or class-switching in germinal centers; indeed, mutations involving MYC, BCL2, and BCL6 identical to those found in lymphomas are also found at a low frequency in normal germinal center B cells obtained from both children and adults. Thus, the relatively high frequency of tumors derived from germinal center B cells likely reflects the error-prone nature of the molecular events that permit antibody class-switching and affinity maturation.
T-Cell Development
Progenitors from the bone marrow that travel to the thymus become committed to T-cell differentiation via interactions with thymic epithelial cells (TEC) (see Fig. 103.1). TECs express ligands for Notch receptors such as DLL4, leading to activation of the receptor NOTCH1, which is essential for early stages of T-cell development.37 As during early B-cell development, early T-cell development is controlled by a transcriptional program induced by a master transcription factor (NOTCH1) and by survival signals mediated by complexes containing components of the T-cell receptor (TCR).38 In most developing T cells, this begins with rearrangement of the TCRβ genes, which (as in B cells) requires RAG1 and RAG2 and involves the participation of TdT. Productive, in-frame rearrangement of the TCRβ gene permits expression of the TCRβ polypeptide, which pairs with pre-Tα polypeptides and assembles into the pre–T-cell receptor. Prosurvival signals transmitted by pre-Tα allow cells to go on to rearrange the TCRα genes, and cells with productive TCRα rearrangements express TCRαβ receptors on their cell surfaces in complex with CD3 polypeptides. Surviving cells also upregulate the CD4 and CD8 coreceptors and proceed through both negative and positive antigenic selection, during which cells expressing autoreactive TCRs or TCRs that fail to recognize antigen in the context of major histocompatibility complex (MHC) antigens are eliminated by apoptosis.39 Cells emerging from the thymus as naïve T cells express either CD4, a coreceptor for MHC class II antigens, or CD8, a coreceptor for MHC class I antigens. A much smaller subset of thymic T-cell progenitors productively rearrange their δ and γ TCR genes, and emerge from the thymus as naïve γδ-TCR–expressing T cells.
Like naïve B cells, naïve T cells home to peripheral tissues under the influence of chemokines, with most γδ T cells homing to gut and skin, and αβ T cells homing much more widely to secondary lymphoid tissues and other sites. γδ T cells are considered to be relative primitive cells that contribute to natural immunity, whereas αβ T cells can differentiate further into a number of different types of effector cells, depending on the dose, timing, and context of subsequent antigenic exposures. αβ T cells recognize antigen when it is presented in the context of an MHC molecule. CD4+ T cells, or T-helper cells, bind to and recognize an antigen presented by MHC class II molecules, whereas CD8+ T cells or cytotoxic T cells bind to and recognize antigen presented by MHC class I molecules. Activation also requires CD40 and CD40L interaction and CD28/CTLA4 and B7 interaction between the T cell and the antigen presenting cell (APC).40 Antigen stimulation of CD8-positive T cells may give rise to CD8-positive effector cytotoxic cells or to long-lived CD8-positive memory cells. By contrast, antigen stimulation of CD4-positive cells can produce a number of CD4-positive effector cell types, including: T helper 1 (Th1) cells, which activate macrophages and cytotoxic T cells through their production of interleukin 2 (IL-2) and interferon gamma; T helper 2 (Th2) cells, which activate B cells through their production of IL-4, -5, -6, and -1341,42; T helper 17 (Th17) cells, which stimulate neutrophils through production of IL-17 and IL-22; and regulatory T cells (Treg), which produce immunosuppressive cytokines such as IL-10. Finally, follicular helper T cells are CD4+ T cells that home to the germinal center via CXCR5 and CXCL13 interactions and play a role in B-cell Ig class switching and Ig production.43
Natural Killer Cells
There is a third class of lymphocytes that can kill targets without MHC restriction, namely NK cells, which are a component of the innate host immune system. NK cells recognize and kill cells that lack MHC class I molecules (including virally infected cells and malignant cells), as well as antibody-coated targets through interactions with Fc receptors on the NK cell surface. NK cells lack surface CD3 and do not have rearranged TCR genes. Morphologically, these cells are slightly larger than resting T and B cells and have paler cytoplasm that contains azurophilic granules, an appearance similar to that of activated cytotoxic T cells.
Immunophenotyping of Lymphoid Cells
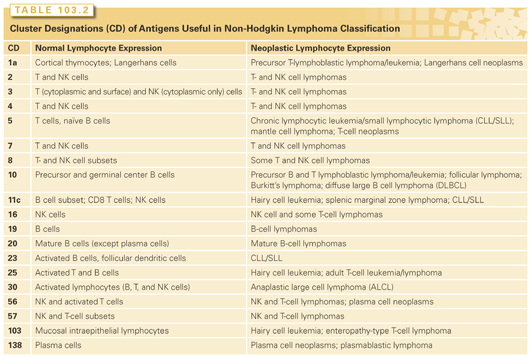
As has been alluded to, lymphocytes at various stages in ontologic development can be defined and differentiated by the detection of certain antigens on the cell surface (see Table 103.2). This antigen footprint is referred to as the immunophenotype of the cell. It can be detected by a flow cytometric analysis of single cell suspensions from whole blood, bone marrow, body fluid, or disaggregated tissue using fluorescently labeled antibodies against these antigens or by immunohistochemistry, which involves the incubation of paraffin-embedded tissue sections with enzyme-linked antibodies against these antigens followed by a colorimetric reaction. These techniques have become vital in diagnosing and monitoring lymphomas, and have provided insight into the normal counterparts of the malignant lymphocyte.
Chromosomal Translocations and Oncogene Rearrangements
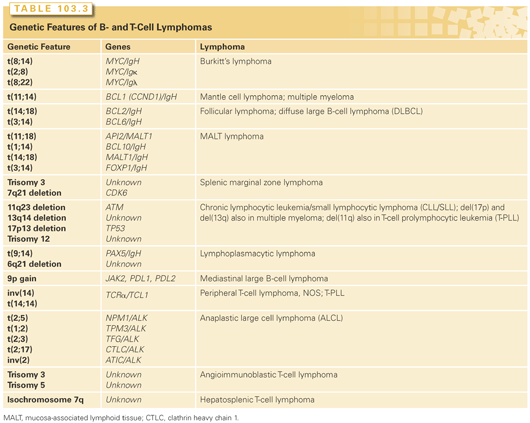
Given the mechanism of Ig and TCR gene rearrangements in lymphoid cells—namely, the formation of DNA breaks with the joining of new pieces of DNA—it is not surprising that lymphomas are frequently found to have chromosomal translocations that involve the activation of an oncogene or inactivation of a tumor suppressor gene (Table 103.3). The former is more common, whereby a proto-oncogene is brought under the control of a constitutively active promoter. The resulting overexpression of the involved gene (now called an oncogene) conveys oncogenic properties on the gene and its protein product, which is responsible for induction and/or maintenance of some aspect of the transformed phenotype. Examples of this type of event include the (8;14)(q24;q32) translocation in BL, involving the MYC proto-oncogene and the IgH gene; the (14;18)(q32;q32) translocation in follicular lymphoma, involving the BCL2 proto-oncogene and the IgH gene; and the (11;14) (q13;q32) translocation in mantle cell lymphoma, involving the gene encoding cyclin D1 (CCDN1) and the IgH gene. Less commonly, chromosomal translocations produce fusion genes that encode chimeric oncogenic proteins. Examples of this include the (2;5)(p23;q35) translocation involving the ALK and NPM1 genes in anaplastic large cell lymphoma (ALCL) and the t(11;18)(q21;q21) translocation involving the API2 and MLT genes in mucosa-associated lymphoid tissue (MALT) lymphoma. These translocations and rearrangements can be detected by polymerase chain reaction (PCR) using probes that span the chromosomal breakpoints, reverse transcriptase PCR (RT-PCR) to detect the RNA product of the fusion gene, or fluorescence in situ hybridization (FISH) using probes to specific chromosomal segments. In cases where the translocation results in the expression of a protein or portion of a protein that is never expressed in normal lymphocytes (e.g., anaplastic lymphoma kinase [ALK] kinase), immunohistochemistry can be used to detect the protein and infer the presence of a rearrangement involving the gene that encodes that protein.
In 2001, the World Health Organization (WHO) published a new classification of tumors of the hematopoietic and lymphoid tissues (Table 103.4).44 It was the end result of a project that began in 1995 with 10 committees of pathologists and a Clinical Advisory Committee of international experts to ensure the clinical utility of the classification system. It incorporated, with minor edits, the 1994 consensus by the International Lymphoma Study Group regarding a list of lymphoid neoplasms that were distinct and recognizable by pathologists called the Revised European and American Lymphoma (REAL) classification.45 The principle behind the classification system is to use and integrate all of the relevant information, including morphology, immunophenotype, genetics, and clinical features, to define disease entities with the relative importance of each type of information varying from disease to disease. The WHO classification system was recently updated and diseases were reclassified in 2008 based on new and evolving information with regard to each of these disease characteristics.
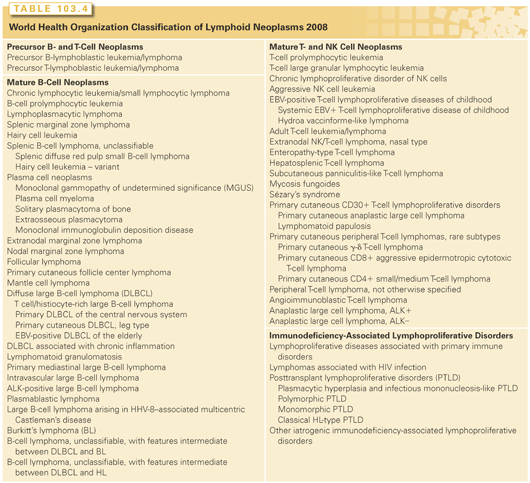
Categories of Lymphoid Neoplasms
There are five main categories of lymphoid neoplasms defined by the WHO: precursor B- and T-cell neoplasms, mature B-cell neoplasms, mature T/NK cell neoplasms, HL, and immunodeficiency-associated lymphoproliferative disorders (see Table 103.4). Each category is a set of distinct diagnoses that are not further classified or grouped by grade, prognosis, or clinical behavior but instead are considered unique entities. In 2001, there were 48 such entities plus additional variants. In 2008, several additions were made, and several provisional diagnostic categories were created that reflect the additional information being gleaned from technology, such as gene expression profiling (GEP) and the consequent recognition of heterogeneity within existing disease entities. For example, within DLBCL, GEP can differentiate between two broad categories of disease, namely the germinal center B-cell type (GCB) and the activated B-cell type (ABC), with different prognoses.46 These categories have not yet been adopted into the WHO because they are not yet relevant to differential treatment strategies, but new treatments are being developed with these GEPs in mind, and treatment effect is being stratified by DLBCL subtype in ongoing clinical trials, and this will likely become clinically relevant in the near future. Furthermore, disease location is being recognized as important in distinguishing one DLBCL from another, with DLBCL of certain locations like the central nervous system (CNS) or primary cutaneous DLBCL, leg type, having unique clinical presentations, clinical behavior, and GEPs compared with nodal DLBCL; these have been added as distinct entities to the 2008 WHO. The microenvironment is another important defining feature of some lymphomas and, as such, T-cell/histiocyte–rich large B-cell lymphoma was added to the updated classification. Finally, two provisional categories have been created to recognize lymphomas that have features intermediate between two types of lymphoma, the so-called gray zone lymphomas: B-cell lymphoma unclassifiable (BCLU) with features intermediate between BL and DLBCL (BCLU-BL/DLBCL) and BCLU with features intermediate between HL and DLBCL (BCLU-HL/DLBCL). These categories were created recognizing that they are likely a heterogenous group of disease, with some most closely resembling BL or HL, some most closely resembling DLBCL, and some belonging to distinct entities, helping to create a more systematic approach to their study and classification.
PRINCIPLES OF MANAGEMENT OF NON-HODGKIN’S LYMPHOMA
Differential Diagnosis and Sites of Disease at Presentation
More than two-thirds of patients with NHL present with persistent painless peripheral lymphadenopathy. At the time of presentation, a differential diagnosis of generalized lymphadenopathy necessitates the exclusion of infectious etiologies such as bacteria (including mycobacteria), viruses (e.g., infectious mononucleosis, cytomegalovirus, hepatitis B, HIV), and parasites (toxoplasmosis) as well as inflammatory and autoimmune diseases, and metastatic malignancies. It is generally agreed that a lymph node larger than 1.5 × 1.5 cm that is not associated with a documented infection and that persists longer than 4 weeks should be considered for a biopsy.47 A biopsy should be performed immediately for patients with other findings suggesting malignancy (e.g., systemic complaints or B symptoms, such as fever, night sweats, weight loss). However, lymph nodes in several histopathologic subtypes of NHLs frequently wax and wane. In teenagers and young adults, infectious mononucleosis and HL should be placed high in the differential diagnosis. Involvement of Waldeyer’s ring, epitrochlear, and mesenteric nodes are more frequently observed in patients with NHL than in patients with HL. About 40% of all patients with NHL present with systemic complaints. B symptoms are more common in patients with aggressive histologies approaching 50%. Less frequent presenting symptoms, occurring in less than 20% of patients, include fatigue, malaise, and pruritus.
NHLs also present with thoracic, abdominal, and/or extranodal symptoms. Although much less common than with HL, approximately 20% of patients with NHL have mediastinal adenopathy. These patients most frequently present with persistent cough, chest discomfort, or without clinical symptoms but have an abnormal chest radiograph. Occasionally, a superior vena cava syndrome accompanies presentation. A differential diagnosis of mediastinal presentation includes infections (e.g., histoplasmosis, tuberculosis, infectious mononucleosis), sarcoidosis, HL, as well as other malignancies. Involvement of retroperitoneal, mesenteric, and pelvic nodes is common in most histologic subtypes of NHL. Unless massive or leading to obstruction, nodal enlargement in these sites often does not produce symptoms. In contrast, patients with an abdominal mass, massive splenomegaly, or primary gastrointestinal (GI) lymphoma present with complaints similar to those caused by other space-occupying lesions. These complaints include chronic pain, abdominal fullness, and early satiety, symptoms associated with visceral obstruction or even acute perforation and GI hemorrhage. Rarely, patients present with symptoms of unexplained anemia. Those with aggressive NHLs can present with primary cutaneous lesions, testicular masses, acute spinal cord compression, solitary bone lesions, and rarely, lymphomatous meningitis. Symptoms of primary NHL of the CNS include headache, lethargy, focal neurologic symptoms, seizures, and paralysis.
When NHL involves an extranodal site, the differential diagnosis is more difficult. NHL uncommonly presents in the lungs as bronchovascular, lymphangitic, nodular, or alveolar patterns of involvement.48 Between 25% and 50% of patients with NHLs present with hepatic infiltration, although relatively few present with large hepatic masses. Of the advanced-stage indolent lymphomas, nearly 75% of patients have microscopic hepatic infiltration at presentation. In contrast, primary hepatic lymphoma is rare and is nearly always an aggressive histology. Primary lymphoma of bone is another uncommon extranodal site, occurring in less than 5% of patients and often presenting as bone pain. Most frequently, lytic lesions are observed on standard radiographs. The most common sites of primary lymphoma of bone include the femur, the pelvis, and the vertebrae. Approximately 5% of NHLs are primary GI lymphomas. These tumors are often associated with hemorrhage, pain, or obstruction. The stomach is most frequently involved, followed by the small intestine, and the colon. Most GI lymphomas are of the diffuse aggressive histologies, specifically DLBCL, mantle cell lymphoma (MCL), and intestinal T-cell lymphoma. The most common site for extranodal MZLs is the stomach. A subset of MCLs presents as multiple intestinal polyposis, which may arise at any site in the GI tract. An uncommon presentation (2% to 14%) of NHL is renal infiltration, and even less common is localized presentation in the prostate, testis, or ovary. The typical histologic subtypes of these sites are DLBCL, BL, and gray zone tumors with features intermediate between DLBCL and BL. Rare sites of primary lymphoma include the orbit, heart, breast, salivary glands, the thyroid, and the adrenal gland.
Diagnosis and Initial Management
After the initial biopsy, a careful history and physical exam should be done to help assess the extent and pace of disease. Attention should be paid to the duration of symptoms and pace of symptomatic progression, whether symptoms associated with a poorer prognosis, such as fevers, night sweats, or unexplained weight loss are present, and to localizing symptoms that may point toward lymphomatous involvement of specific sites, such as the chest, abdomen, or CNS. Concurrent illness that may impact therapy or monitoring on therapy should be ascertained, including a history of diabetes or congestive heart failure. A physical exam should pay close attention to all the peripherally accessible sites of lymph nodes; the liver and spleen size; Waldeyer’s ring; whether there is a pleural or pericardial effusion or abdominal ascites; whether there is an abdominal, testicular or breast mass; and whether there is cutaneous involvement because all of these findings may influence further evaluation and disease management.
Laboratory studies should be obtained, including complete blood count, routine chemistries, liver function tests, and serum protein electrophoresis to document the presence of circulating monoclonal paraproteins. The serum beta-2 microglobulin level and serum lactate dehydrogenase (LDH) are important independent prognostic factors in NHL. A bone marrow biopsy should be considered for staging and prognostic purposes depending on the disease histology and the results of other laboratory and staging studies. An evaluation of the cerebrospinal fluid (CSF) for lymphomatous involvement may be indicated in the setting of concerning neurologic signs or symptoms or a disease that has a high propensity to spread to the CNS. The latter includes a disease involving the paranasal sinuses, testes, and epidural space, as well as highly aggressive histologies like BL.
Imaging studies depend on the histology of the lymphoma as well as the clinical presentation. Chest, abdominal, and pelvic computed tomography (CT) scans are essential for accurate staging to assess lymphadenopathy for indolent lymphomas. Radionuclide scans have clinical utility as diagnostic and monitoring studies. 67Gallium scanning, used based on the ability of this isotope to bind transferrin receptors on tumor cells, has been replaced by positron-emission tomography (PET) using 18F-fluorodeoxyglucose (FDG). FDG-PET scanning is highly sensitive for detecting both nodal and extranodal sites involved by NHL. PET scanning is particularly useful for the histologically aggressive lymphomas, including BL, DLBCL, plasmablastic lymphoma, and the aggressive T-cell lymphomas, but is less reliable in lower grade histologies like MZLs.49 The intensity of FDG avidity, or standardized uptake value (SUV), correlates with histologic aggressiveness.50,51 PET scanning detects an actively metabolizing tumor in residual masses following or during chemotherapy, and persistent abnormal uptake predicts for early relapse and/or reduced survival.52 It is more accurate than the detection of a residual mass on CT scans, which can often be a false positive. Consensus recommendations regarding PET scanning were published as a result of an International Harmonization Project. Among the recommendations are that PET only be used for DLBCL and HL, scanning during therapy be only part of clinical trials, and the scan after all therapy is completed should be done at least 3 but preferably 6 to 8 weeks after chemotherapy and 8 to 12 weeks after radiation or chemoradiotherapy. There is no evidence that a long-term follow up should include PET scanning.53 Finally, magnetic resonance imaging (MRI) is useful in detecting bone, bone marrow, and CNS disease in the brain and spinal cord.
Staging and Prognostic Systems
The Ann Arbor staging system developed in 1971 for HL was adapted for staging NHLs (Table 103.5).54 This staging system focuses on the number of tumor sites (nodal and extranodal), the location, and the presence or absence of systemic, or B, symptoms. Table 103.5 summarizes the essential features of the Ann Arbor system.

The concept of staging has less impact in NHL than in HL. Only a minority of patients with both indolent and aggressive NHL have localized disease at diagnosis, and there is little therapeutic benefit to distinguish between stage III and stage IV disease because the treatment options are identical. The prognosis is more dependent on histology and clinical parameters than the stage at presentation. Staging in NHLs, therefore, is done to identify the minority of patients who can be treated with local therapy or combined modality treatment and to stratify within histologic subtypes to determine the prognosis and to assess the impact of treatment.
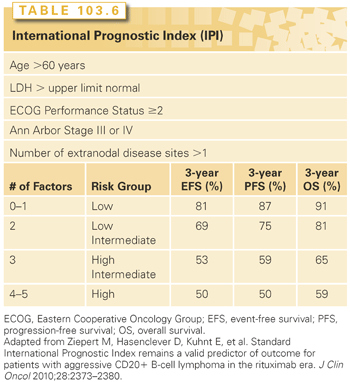
Probably more important than staging is the International Prognostic Index (IPI), which provides risk stratification (Table 103.6).55 The IPI was developed based on an analysis of over 2,000 patients with diffuse aggressive NHLs treated with an anthracycline-containing regimen. This analysis identified age (≤60 years versus >60 years); serum LDH (≤ normal versus > normal); performance status (0 or 1 versus 2 to 4); stage (I or II versus III or IV); and extranodal involvement (≤ one site versus > one site) to be independently prognostic for overall survival. Four risk groups were identified based on the number of risk factors: low risk (0 or 1); low intermediate (2); high intermediate (3); and high (4 to 5). The 5-year overall survival rates for patients with scores of 0 to 1, 2, 3, and 4 to 5 were 73%, 51%, 43%, and 26%, respectively. For the patients aged 60 years or less, only stage, LDH, and performance status were of prognostic significance. Patients ≤60 years with zero, one, two, or three risk factors had 5-year survival rates of 83%, 69%, 46%, and 32%, respectively. Survival rates for those age >60 years with the same scores were 56%, 44%, 37%, and 21%, respectively. The IPI has been adapted following treatment with cyclophosphamide, adriamycin, vincristine, and prednisone plus rituximab (CHOP-R) therapy for DLBCL. Within that model, the 4-year progression-free survival is 94%, 80%, and 53% for zero and one, two, or three or more risk factors, respectively.56
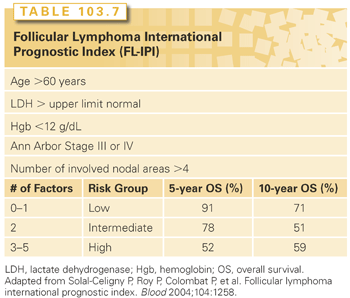
A similar predictive model has been developed for follicular lymphoma based on the analysis of over 4,000 patients with follicular NHL, known as the follicular lymphoma IPI or FLIPI (Table 103.7).57 This study identified the following prognostic factors: age >60 years, stage III/IV, more than four nodal sites, elevated serum LDH concentration, and hemoglobin less than 12. The 10-year survival rates for patients with zero to one (low risk), two (intermediate risk), or three or more (high risk) of these adverse factors averaged 71%, 51%, and 36%, respectively. Similar disease-specific IPIs have been developed for mantle cell lymphoma and peripheral T-cell lymphoma as well. These prognostic indices take into account the proliferative index and cell surface markers, respectively.58,59
More recently, as discussed in the section on the 2008 update to the WHO, GEP has been used to examine DLBCL to identify patients with different prognoses.46 Based on gene expression, DLBCLs have been subclassified into GCB or ABC types. Patients with GCB-like DLBCL had significantly better overall survival than those with the ABC-like variant. Based on findings from GEP, immunohistochemical staining of a limited number of proteins has been proposed as an alternative method for subtyping of DLBCL and prognostication.60 Germinal center and nongerminal center B-cell derivation can be determined by the expression of markers such as CD10, B cell lymphoma–6 protein (BCL-6), and multiple myeloma oncogene 1 (MUM1). Based on immunohistochemistry, it is estimated that approximately 40% of DLBCLs are of the GCB subtype, with the remainder falling into the non-GCB group.
Restaging after treatment is typically done 6 to 8 weeks following the completion of chemotherapy (or chemoimmunotherapy), or 8 to 12 weeks after the completion of radiotherapy or combination chemotherapy and radiotherapy, to assess for disease response to treatment. The most important prognostic factor is the achievement of a complete response to therapy. Restaging at the completion of treatment is often with the repetition of studies that were abnormal at diagnosis. It should be noted that patients with certain lymphomas or bulky disease may not have complete regression of their lymphadenopathy despite there not being any remaining active lymphoma. Nuclear studies, like PET/CT scans, and/or rebiopsy can be helpful in differentiating residual fibrotic tissue from active lymphoma.
Precursor B-Cell and T-Cell Leukemia/Lymphoma
Lymphoblastic lymphoma and acute lymphoblastic leukemia appear to be different manifestations of the same disease entity (see Chapter 110). Cytologically, both are composed of blasts with a high nuclear-to-cytoplasmic ratio, scant cytoplasm, and nuclei with slightly coarse chromatin with multiple small nucleoli. The nuclei may be oval, but more often are folded or convoluted. The blasts are usually intermediate in size, but they may be large, or in unusual cases, so small that there may be confusion morphologically with CLL. When lymph nodes are involved, they are diffusely effaced by blasts. Mitotic figures are usually frequent, and (as with all high-grade lymphomas) some cases contain frequent tingible body macrophages, producing a starry-sky appearance that mimics BL.
Approximately 85% to 90% of lymphoblastic lymphomas are of the T-cell lineage, with the remainder being of the B-cell type. Both are comprised of tumor cells with immunophenotypes that correspond to stages of pre-T and pre-B–cell development, respectively. B-lymphoblastic tumors express CD19 and are variably positive for other B lineage markers and negative in most cases for surface immunoglobulin. T lymphoblastic tumors usually express cytoplasmic CD3 but may be surface CD3 negative, and show variable expression of other T-cell markers. Most lymphoblastic tumors are positive for TdT, a specific marker of immature lymphoid cells that can be detected by flow cytometry or immunohistochemistry.
Although lymphoblastic lymphomas represent a major subgroup of childhood NHLs, they are unusual in adults (2% of adult NHLs). Patients are usually adolescent or young adult males who present with lymphadenopathy in cervical, supraclavicular, and axillary regions (50%) or with a mediastinal mass (50% to 75%). These masses can be associated with superior vena cava syndrome, tracheal obstruction, and pericardial effusions. Less commonly, patients present with extranodal disease (skin, testicular, or bony involvement). More than 80% of patients present with stage III or stage IV disease, almost 50% have B symptoms, and the majority has an elevated LDH. Although the bone marrow can be uninvolved at presentation, virtually all patients develop bone marrow infiltration and a subsequent leukemic phase indistinguishable from T-cell acute lymphoblastic leukemia. Patients with bone marrow involvement have a very high incidence of CNS infiltration. B-cell lymphoblastic lymphoma is a very rare entity, with patients having a median age of 39 years.61 B-cell lymphoblastic lymphomas present without a mediastinal mass but instead involve lymph nodes and extranodal sites.
The treatment of precursor B-cell and T-cell lymphoblastic leukemia/lymphoma is detailed in Chapter 110.
Follicular Lymphoma
Introduction
FL is the second most common lymphoma diagnosed in the United States and western Europe, making up approximately 20% of all NHLs, and 70% of indolent lymphomas.62 The median age at diagnosis is 60 years, and there is a slight female predominance.63,64 The incidence is increased among relatives of persons with FL.65
Pathology
FLs are malignant counterparts of normal germinal center B cells.44 FL recapitulates the architecture of normal germinal centers (GC) of secondary lymphoid follicles.44 The neoplastic cells consist of a mixture of centrocytes (small- to medium-sized cells with irregular or cleaved nuclei and scant cytoplasm) and centroblast (large cells with oval nuclei, several nucleoli, and moderate amounts of cytoplasm). The clinical aggressiveness of the tumor correlates with the number of centroblasts that are present. The WHO classification44 adopted grading from 1 to 3 based on the number of centroblasts counted per high power field (hpf): Grade I, 0 to 5 centroblasts/hpf; Grade II, 6 to 15 centroblasts/hpf; Grade III, more than 15 centroblasts/hpf. Grade III has been subdivided into grade IIIa, in which centrocytes predominate, and grade IIIb, in which there are sheets of centroblasts.66 Although the grading system remains in place, clinically, grade I and II and many cases of grade IIIa FLs are approached similarly. Akin to normal GCs, small numbers of T cells and follicular dendritic cells are present in the malignant follicles; however, tingible body macrophages, cells that have ingested apoptotic cells that are common in reactive GCs, are not observed. Involvement of the peripheral blood with malignant cells is commonly seen, and morphologically, these cells have notches and have been referred to as buttock cells. FL grade IIIb is an aggressive disease grouped with diffuse large B-cell lymphoma. Bone marrow involvement is exceedingly common in FL patients, usually taking the form of paratrabecular lymphoid aggregates.44
Immunophenotype and Genetics
FL cells express monoclonal immunoglobulin light chain, CD19, CD20, CD10, and BCL6 and are negative for CD5 and CD23. In virtually all cases, FL cells overexpress BCL-2. Clonal Ig gene rearrangements are present and, in most cases, the Ig loci have extensive somatic mutations, further supporting a GC origin. Approximately 85% of FLs have the t(14;18), which drives overexpression of BCL-2, a member of a family of proteins that blocks apoptosis. However, multiple genetic events are required for the development of FL, because the t(14;18) can be identified in a small fraction of normal B cells in most normal children and adults. Deep sequencing studies have established that the most common mutations in FL (90% of tumors) involve mixed-lineage leukemia 2 protein (MLL2), a gene encoding a histone H3 methylase.67,68 Less common recurrent mutations involve other genes involving epigenetic modifying genes, such as EZH2, CREBBP, and EP300, indicating that genetically determined alterations in the epigenome contribute to FL in ways that remain to be defined. Other recent studies suggest that reactive cells within the malignant microenvironment also contribute to the pathobiology of FL, based on evidence that immune signatures of T cell and macrophage infiltration defined by gene expression profiling are predictive of outcome.69
Clinical Features
Patients with FL generally present with asymptomatic lymphadenopathy, which often waxes and wanes over the course of years. Bone marrow involvement is present in 70% of patients, whereas involvement of other nonlymphoid organs is uncommon. Less than 20% of patients present with B symptoms or an increased serum LDH. In a small subset of patients, the disease presents in the intestine; such patients usually have an early stage and a favorable prognosis.70 Histologic transformation of FL to DLBCL occurs in 10% to 70% of patients over time, with a risk of about 2% to 3% per year71–73 and is associated with the rapid progression of lymphadenopathy, extranodal disease (besides the marrow), B symptoms, elevated serum LDH, and calcium.
Prognosis
Measures of outcome include the FLIPI (see Table 103.7) and tumor grade.74 A modified version of this score, the FLIPI2, evaluated five parameters, with some overlap of the FLIPI.75 The utility of the FLIPI2 model remains uncertain. Since the incorporation of rituximab into the mainstream therapy of FL, the FLIPI has continued to be a useful prognostic model.76
FL tumors are graded from 1 to 3 and this grade has some prognostic utility. There has generally been suboptimal consensus of pathologists on grading FL. There is no evidence to support a different treatment approach between grade I and grade II FL. Differences in molecular genetics as well as clinical behavior suggest that FL grade IIIa is more commonly an indolent disease, whereas grade IIIb is an aggressive disease.44,77
The investigation of the cellular microenvironment of FL has provided interesting insights into prognosis.78–87 It has been suggested that FL is an immunologically functional disease in which an interaction between the tumor cells and the microenvironment modulates clinical behavior. These studies, which have observed an impact on the prognosis of reactive macrophages and T cells, need additional study in larger data sets and a prospective design with uniformly treated patient populations.
Treatment of Early Stage Disease
Less than 10% of patients with FL have stage I/II disease.88 Radiation therapy is the treatment of choice for limited stage FL and results in a 5-, 10-, and 15-year freedom from treatment failure of 72%, 46%, and 39%, and an overall 5-, 10-, and 15-year survival rates of 93%, 75%, and 62%, respectively, with a median survival of approximately 19 years.89 A dose of 24 to 30 Gy appears to be highly effective, with no evidence of benefit for higher doses.90 However, most patients with stage I disease treated in the United States do not receive radiation therapy.88 This is surprising given a large study of over 6,000 patients with stage I or stage II FL diagnosed from 1973 to 2004, 34% of whom were initially treated with RT, where patients who received initial radiation therapy (RT) had higher rates of disease-specific survival at 5 years (90% versus 81%), 10 years (79% versus 66%), 15 years (68% versus 57%), and 20 years (63% versus 51%).91
In pre-rituximab era studies, adjuvant chemotherapy probably does not add additional benefit after local RT.92 A recent retrospective analysis suggested an improved progression-free survival (PFS) outcome with chemoimmunotherapy or systemic therapy plus RT as compared to RT alone, with no impact on overall survival (OS).93 This will require additional study. If significant morbidity is possible from RT based on the location of the disease area or if the patient chooses to not receive RT, observation may be a reasonable alternative, especially for stage II patients.94 In this report, the median OS of selected untreated patients was 19 years. At a median follow-up of 7 years, 63% of patients had not required treatment.
Treatment of Advanced Stage Disease
The overwhelming majority of patients have advanced stage disease at diagnosis. Patients with asymptomatic FL do not require immediate treatment unless they have symptomatic nodal disease, compromised end organ function, B symptoms, symptomatic extranodal disease, or cytopenias. This approach is supported by randomized prospective trials of observation versus immediate treatment. One of the largest trials compared immediate treatment with chlorambucil to observation.95 At a median follow-up of 16 years, no difference in OS and cause-specific survival was seen between the two approaches. Similar results have been noted in other prospective trials of initial treatment versus observation.96
A major question is whether rituximab might change this approach in early treatment in asymptomatic patients. A retrospective analysis of good risk patients who were either observed or received single-agent rituximab97 found no negative impact of watchful waiting. A prospective study compared observation to rituximab alone or rituximab followed by maintenance in previously untreated FL. The median time to next treatment was 34 months in the watch and wait patient but was not reached in the rituximab-treatment arm. The 3-year PFS was 33%, 80%, and 90% of the observed, rituximab, or rituximab followed by maintenance patients, respectively, with 95% OS in all three groups. The important issues of time to second therapy, quality of life, impact on histologic transformation, cost, toxicity, and future responses to rituximab are not yet addressed.98
Rituximab has changed the paradigm of treating FL. The recent improvement in survival of patients with FL is largely due to the use of anti-CD20 monoclonal antibody-based therapy.99 The benefit of adding rituximab to combination chemotherapy for the initial treatment has been demonstrated in multiple randomized trials of chemotherapy with or without rituximab (see Table 103.2).100–104 All of these trials have demonstrated improved response rates and time to progression in the rituximab plus chemotherapy arms, as well as improvement in OS. FDG-PET scanning has been employed to evaluate responses to CHOP-R in previously untreated patients. PET scanning was predictive when performed after four cycles and at the end of therapy. The 2-year PFS was significantly higher for PET-negative than PET-positive patients when employed as an interim or end of therapy scan. The 2-year OS was also significantly higher for PET-negative than PET-positive patients. This will require further study but may change management in the future.105
Other chemotherapy drugs plus rituximab have also been used for the initial therapy of FL. Bendamustine plus rituximab (BR) has been compared to CHOP-R in a randomized phase III trial with bendamustine (90 mg/m2 days 1 and 2) plus rituximab (375 mg/m2 day 1) in 513 patients with advanced follicular, marginal zone, lymphoplasmacytic, and mantle cell lymphoma.106 In this study, a superior median PFS in favor of BR versus CHOP-R was seen (69.5 versus 31.2 months) at 45 months. Moreover with BR, less toxicity, including lower rates of grade 3 and 4 neutropenia and leukopenia were observed. There was no difference in OS at a median follow-up of 45 months. Intensifying the schedule of CHOP-R from every 21 to every 14 days was also of no benefit.107 Fludarabine plus rituximab108 and fludarabine, mitoxantrone, dexamethasone, and rituximab109 both showed response rates of over 90% in previously untreated patients. However, significant neutropenia and opportunistic infections were observed with these regimens. A randomized phase III trial compared three regimens in previously untreated stage II to IV FL patients: CHOP-R; cyclophosphamide, vincristine, prednisone, and rituximab (CVP-R); or rituximab, fludarabine, and mitoxantrone (R-FM). Both R-FM and CHOP-R were superior to CVP-R in 3-year PFS and time to treatment failure (TTF), but there was no difference in OS.110 The current impact of this study is uncertain given the favorable results and lower toxicity seen with BR.
Rituximab alone has been used as the first therapy in patients with FL, with overall response rates of around 70% and complete response (CR) rates of over 30% reported.111–113 The most favorable data of single-agent rituximab is the recent update of the swiss group for clinical cancer research (SAKK) trial.114 Patients received four weekly doses, and then patients with stable disease or better were randomized to observation or four doses of maintenance therapy, one dose every 2 months. In this study, 202 patients with previously untreated or relapsed/refractory FL administered four weekly doses of single-agent rituximab has been reported. The 151 patients with responding or stable disease at week 12 were randomized to no further treatment or prolonged rituximab maintenance every 2 months for four doses. At a median follow-up of 35 months, patients who received the prolonged rituximab maintenance had a twofold increase in event-free-survival (23 months versus 12 months). With a longer follow-up, 45% of newly diagnosed patients in this study were in remission at 8 years with the addition of maintenance rituximab.
Maintenance rituximab has also been shown to benefit patients who received chemotherapy without rituximab as part of the initial treatment. A randomized trial of maintenance rituximab versus observation after CVP with the majority having FL reported that patients who received maintenance rituximab had improved rates of 3-year PFS (68% versus 33%). Survival rates were similar between the two groups.115 With the current paradigm of treating patients with concurrent chemotherapy plus rituximab, this study has less applicability.
The use of maintenance rituximab after chemoimmunotherapy in patients with FL has been examined in a large randomized trial.116 Although maintenance rituximab appears to improve PFS rates, toxicities, albeit tolerable, are increased and the effect on OS is, to date, unclear. The Primary Rituximab and Maintenance (PRIMA) phase III intergroup trial randomly assigned 1,018 patients with previously untreated FL that responded to chemoimmunotherapy (CVP-R, CHOP-R, or fludarabine, cyclophosphamide, mitoxantrone, and rituximab [FCM-R]) maintenance with rituximab (375 mg/m2 every 8 weeks for 24 months) or placebo.116 At a median follow-up of 36 months from randomization, patients assigned to rituximab maintenance had a higher rate of PFS (75% versus 58%). A higher percentage of patients in complete response/complete response, unconfirmed (CRu) at 24 months (72% versus 52%) was also seen 2 years postrandomization in patients receiving maintenance rituximab. There was a significantly higher percentage of patients with grade III/IV adverse events and infections in the rituximab maintenance group. At this time, OS is the same in both groups.
Radioimmunotherapy alone has been used as the initial treatment in a limited number of patients with FL. 131Itositumomab was given to 76 previously untreated patients with FL leading to overall and complete response rates of 95% and 75%, respectively, and, at 5-years, OS and PFS rates of 89% and 59%, respectively.117 131I tositumomab is no longer commercially available. 90Yttrium (90Yi)-ibritumomab tiuxetan has also been studied as sole initial therapy with excellent results with limited follow-up.118
Radioimmunotherapy has also been used as consolidation following conventional chemotherapy induction in patients with FL. Both 90Yi-ibritumomab tiuxetan and 131I-tositumomab have been studied. This approach has been associated with very high response rates, conversions of partial response (PR) to CR, and well-maintained responses.119–121 A phase III trial compared 90Yi-ibritumomab tiuxetan to observation following a CR or PR to induction chemotherapy for treatment-naïve patients with FL.122 Of note, the majority of patients did not receive rituximab along with the induction chemotherapy. At 8 years, both the PR and CR patients who received 90Yi-ibritumomab tiuxetan had significantly longer median PFS with improvement of about 36 months. In contrast to this study, a randomized trial of CHOP plus rituximab to CHOP followed by 131Itositumomab did not see any differences in PFS between the two arms.123
High-dose therapy and autologous stem cell transplantation (ASCT) has been used to consolidate first remission for patients with FL. These studies generally preceded the widespread use of rituximab. With ASCT in first remission, about 50% of patients are disease free at 10 years and beyond following ASCT, but an increased risk of second malignancies, including myelodysplastic syndrome (MDS), acute myelogenous leukemia (AML), and solid tumors, has been observed with long follow-ups of these patients. Several randomized trials have examined the role of ASCT in previously untreated patients with FL following an induction therapy.124–129 The majority of these studies have demonstrated a significant improvement in PFS, but no impact on OS.130 One reason for the lack of impact on OS has been the excess number of second malignancies.
Although allogeneic stem cell transplantation (alloSCT) can potentially lead to a cure for patients with FL due to the significant treatment related mortality, this is largely reserved for patients with relapsed and more refractory disease.
Treatment of Relapsed FL
When patients with relapsed FL require treatment, there are many options, ranging from rituximab alone to combination chemotherapy plus rituximab, radioimmunotherapy, and for selected patients, stem cell transplantation.
A recent update of single-agent rituximab therapy in patients with relapsed FL is from the randomized SAKK trial.114 With a long follow-up, 35% of responders remain in remission at 8 years. However, in the context of current induction therapy that includes chemotherapy and rituximab in the majority of patients, it is uncertain if the response data to single-agent rituximab is as high or durable as in patients who received chemotherapy without rituximab as induction therapy. There is a evidence, however, that retreatment with rituximab in patients with relapsed, largely FL, who had previously responded to rituximab had a response rate of 40% with a median time to progression of 18 months following retreatment.131
The combination of chemotherapy and rituximab has enhanced the efficacy of treatment of relapsed FL. Probably the largest study treated selected patients with relapsed FL who were previously not treated with an anthracycline- or rituximab-containing regimen.132 Patients were randomized to CHOP or CHOP-R and responding patients were randomized to 2 years of maintenance rituximab or observation. The overall and CR rates were significantly improved in the CHOP-R group, and the median PFS was improved by approximately 12 months. An update of this study with a median follow-up of 6 years reported that maintenance rituximab also improved median PFS by 2.4 years. The OS at 5 years following maintenance was 74% versus 64% with observation alone. Given the current paradigm of chemoimmunotherapy and maintenance, the applicability of these data to presently treated patients is uncertain.
Another regimen in which a benefit for the addition of rituximab was seen for relapsed disease in a randomized trial employing FCM.133 A number of phase 2 trials of other agents plus rituximab associated with quite high response rates included BR with 90% response rate (RR) and median PFS of 2 years.134–136 Single-agent bendamustine has an overall RR of 77% with a median response duration of 6.7 months.134 With more widespread use of BR as initial therapy, BR will be employed less for recurrent disease. The regimen FCR has a similarly high response rate but with significant myelosuppression.137 Phase 2 studies employing bortezomib and rituximab and bortezomib, rituximab, and bendamustine have reported RRs of approximately 50% and 93%, respectively.138,139
The anti-CD20 radioimmunotherapy agents have been employed for treatment of patients with relapsed and refractory FL.140 The RRs in this patient population are similar with both agents, with 60% to 80% of patients responding. The median PFS is about 12 months, although the approximately 20% to 37% of patients who achieve a CR have a median time to progression of approximately 4 years.141,142 A randomized trial compared single-agent rituximab to 90Yi-ibritumomab in patients with relapsed indolent (predominantly FL).143 The overall and CR rates were significantly higher with radioimmunotherapy (RIT), but no difference in time to progression or OS was observed. Retreatment with these agents remains controversial, with uncertainty of delivery of full dose and concerns of second malignancies.144
FL is extremely responsive to RT; low-dose RT (e.g., total dose of 4 Gy, given as two consecutive daily 2-Gy fractions) can be used for the palliation of patients who have symptoms related to a single disease site, with CR rates of 57% and overall RRs of 82%.145 Patients who go into CR have long, durable local control rates. There are no significant side effects of treatment, even in the head and neck region where higher doses would cause xerostomia and mucositis.
The use of either ASCT or alloSCT in FL is controversial and the subject of numerous clinical trials.146 A large number of phase 2 studies prior to the availability of rituximab, involving high-dose therapy and autologous hematopoietic stem cell transplantation (HCT) have shown that approximately 40% of patients with good performance status and chemosensitive relapsed disease may experience prolonged PFS and OS.147–151 Prior to the widespread use of rituximab for in vivo purging, many strategies were taken to render the autologous stem cell collections free of lymphoma cells. Although single institution studies suggested that reinfusion of tumor-free stem cells led to a decreased relapse rate, it remains controversial as to whether there is a benefit, particularly now with rituximab treatment. The only phase 3 randomized trial (the chemotherapy, unpurged stem cell transplantation, purged stem cell transplantation [CUP] trial) comparing ASCT to conventional chemotherapy in relapsed FL patients demonstrated a higher PFS and OS for ASCT, and no benefit for purging the stem cell graft.152 A retrospective analysis of patients undergoing ASCT following rituximab-based salvage therapy did not suggest a benefit of ASCT as compared to conventional therapy. Unfortunately, as has been seen in ASCT in first remission, second malignancies—both solid tumors and MDS and AML—are reported following ASCT.
Phase 2 studies have looked at the use of in vivo purging pre-ASCT and maintenance therapy with rituximab following ASCT in patients with relapsed FL. These suggest an improvement in PFS, similar to what has been seen following conventional chemotherapy and chemoimmunotherapy. A phase 3 trial in patients with relapsed FL has investigated the inclusion of rituximab for in vivo purging pre-ASCT and 2 years of maintenance post-ASCT.153 There was an improvement in PFS for patients receiving rituximab for in vivo purging, maintenance, and the combination of both as compared to no rituximab, but no OS benefit.
AlloSCT has been investigated in patients with relapsed FL. Both myeloablative and reduced intensity conditioning (RIC) approaches have been employed. Unfortunately, myeloablative conditioning has a treatment related mortality of up to 40%; however, the relapse rate is less than 20%.154 There is enthusiasm for RIC alloSCT because it has lower treatment-related mortality,155–157 but some reports suggest that the relapse rate may be higher than conventional myeloablative conditioning. The role of alloSCT versus ASCT for FL remains uncertain. A recent National Comprehensive Cancer Network (NCCN) database retrospective analysis found significantly higher 3-year OS for ASCT versus alloSCT (87% versus 61%).158 Certainly, for younger patients with more resistant disease, alloSCT remains a potentially curative option for relapsed FL.
Histologic Transformation
Part of the natural history of any indolent B-cell NHLs is progression to a higher grade histologic subtype, most commonly DLBCL, but much less commonly, BL or even HL can be seen.71,159 Histologic transformation (HT) is most commonly seen in FL, but is also seen in patients with MZL, lymphoplasmacytic lymphoma, and small lymphocytic lymphoma/CLL (where this is referred to as a Richter transformation), and a biopsy is critical in order to demonstrate transformation. HT occurs at a rate of approximately 2% to 3% per year.72 The clinical presentation of HT includes rapid growing masses, extranodal disease, B symptoms, hypercalcemia, and elevated serum LDH. TP53 mutations and translocations or amplifications of MYC are the most common genetic abnormalities seen in HT.
Historically, HT to DLBCL has been associated with a very poor prognosis. In a series from Stanford, previously untreated patients and patients with limited disease and no prior therapy at transformation had improved prognoses.160 Although the median survival for all patients with transformation was only 22 months, those who achieved a CR to combination chemotherapy had an actuarial survival of 75% at 5 years. More recent studies suggest that CHOP-R may improve OS for patients with transformed disease. Patients who have not previously received an anthracycline-containing regimen should be treated with CHOP-R and, assuming a CR is obtained, monitored. For previously treated patients, high-dose therapy and ASCT should be considered assuming the patient has chemosensitive disease. Patients with histologic transformation can have later relapses with indolent lymphoma.
Newer Agents
There are a multitude of new approaches that have been studied in patients with FL. This includes monoclonal antibodies, idiotype vaccines, immunomodulatory agents, and novel drugs such as kinase inhibitors.
Monoclonal antibodies directed against other B-cell–associated antigens as well as new anti-CD20 monoclonal antibodies (mAb) are being investigated in FL. These have included anti-CD80,161,162 anti-CD22 mAbs,163,164 and anti-CD40.165 Several new anti-CD20 monoclonal antibodies are being evaluated in patients with FL who are refractory to rituximab. These include several humanized antibodies that are designed to have less infusion toxicity and a better antibody-dependent cell-mediated cytotoxicity effector function.166–168 The other mAb of interest is obinutuzumab, the first type II, glycoengineered, and humanized monoclonal anti-CD20 antibody.169 In rituximab-refractory patients in the high-dose cohort, the RR was 55% with a median PFS of 11.9 months. Studies of obinutuzumab in combination with chemotherapy have shown 93% to 98% RRs in relapsed and refractory FL patients.170
A number of immunostimulatory agents have been studied to enhance the activity of rituximab. These include cytokines such as IL-2 and immunostimulatory DNA sequences known as CpGs.171 To date, although having immunomodulatory effects, the impact on enhancing the therapeutic effect of rituximab has been limited. A phase 2 study of lenalidomide plus rituximab has reported high RRS, but a phase 3 study will be needed to demonstrate superiority over rituximab alone.172
The other area of interest has been in active immunization, focusing largely on the idiotype protein as the antigen. To date, there have been three randomized studies employing idiotype proteins coupled to a protein called keyhole limpet hemocyanin (KLH) following the induction of remission in patients with FL. The Favrille trial used rituximab for induction therapy. The median time to progression (TTP) was 9 months for the idiotype-KLH (Id-KLH) vaccinated patients and 12.6 months in the control group (p = 0.019).173 However, this difference was attributed to more patients with high-risk FLIPI scores in the Id-KLH arm. The Biovax study reported showing a 14-month improvement in PFS for the Id-KLH vaccinated patients as compared to control; however, the induction chemotherapy was intense and remissions had to be sustained for 12 months prior to the initiation of vaccination.174 The trial using the MyVax Id-KLH conjugate following CVP chemotherapy failed to show any PFS advantage. Based on these studies, it is unlikely at the present time that idiotype vaccinations will be pursued in FL.
B-cell kinases are logical targets for therapy in FL. To date, three kinase inhibitors, idelalisib, ibrutinib, and fostamatinib, which target the phosphoinositide 3-kinase (PI3k) p110δ, Bruton’s tyrosine kinase (BTK), and spleen tyrosine kinase (SYK), respectively, have been tested. In relapsed and refractory FL patients, the response rates to idelalisib,175 ibrutinib,176 and fostamatinib177 were 62% (including other indolent NHLs besides FL), 27%, and 10%, respectively. These agents are undergoing additional study, in combination with chemotherapy and as maintenance following remission induction, to better define their role.
Follicular Lymphoma Grade III
FL grade III has been historically referred to as follicular large cell lymphoma. It is histologically defined by the presence of more than 15 centroblasts per hpf. It is further subdivided into grade IIIa, where centrocytes are present, and grade IIIb, where there are sheets of centroblasts. These are further differentiated by the presence of BCL6 rearrangements in a high fraction of grade IIIb cases. Because many studies likely include both grade IIIa and IIIB, this heterogeneity may affect an interpretation of the outcomes. Although the follicular architecture is intact, the clinical presentation, behavior, and outcome with treatment in many patients with FL grade IIIb more closely approximates that of DLBCL.178–180 In contrast to DLBCL, the relapse rate of FL grade IIIb is higher in some series, but survival is longer.181 A recent series suggested similar outcomes of grade IIIa and IIIB cases and no benefit for the inclusion of anthracyclines in the treatment regimen.182
Small Lymphocytic Lymphoma/B-Cell Chronic Lymphocytic Leukemia
Introduction
Small lymphocytic lymphoma (SLL) is a mature (peripheral) B-cell malignancy. It is synonymous with CLL. The malignant cells in SLL and CLL are morphologically, immunophenotypically, and genetically identical. The difference between these two diagnoses is the clinical presentation, with a nonleukemic presentation in SLL. The diagnosis is made by an examination of involved tissue, such as the lymph node or bone marrow.
SLL represents less than 5% of all NHLs. CLL/SLL comprises 90% of chronic lymphocytic leukemias in Western countries. Less than 10% of patients present with only nodal involvement (i.e., SLL). However, most patients with SLL at presentation ultimately develop bone marrow and blood infiltration. The median age at diagnosis is 65 years.63 At least 80% have stage IV disease due to bone marrow involvement at diagnosis.
Pathology
The cells within lymphoid tissues in CLL/SLL are small lymphocytes with condensed chromatin, round nuclei, and occasionally, a small nucleolus.44 Larger lymphoid cells with prominent nucleoli and dispersed chromatin are also seen. These larger lymphoid cells are usually clustered together in so-called proliferation centers, which are pathognomonic. Roughly 60% of SLL/CLLs have Ig genes that show evidence of significant somatic mutation, defined as a rearranged Ig heavy-chain gene with a sequence that differs from germ-line position at 2% or more of the Ig V region nucleotides, which is taken as evidence of origin from an antigen-stimulated B cell.183
Immunophenotype and Genetics
SLL/CLL cells express low-level monoclonal surface Ig, usually IgM or IgM and IgD. They also express human leukocyte antigen-DR (HLA-DR) and the B-cell antigens CD19, CD20, and CD23, and are characteristically CD5 positive. About 40% of cases express CD38. Expression of the tyrosine kinase ZAP70 is also observed in a subset of cases and correlates with a more aggressive clinical course.184
Immunoglobulin genes are clonally rearranged, with IgV region somatic mutations in up to 60% of patients. Cytogenetic abnormalities include trisomy 12, which is present in about 40% of cases, as well as 13q deletions (45% to 55% of cases), 11q deletions (17% to 20% of cases), and 17p deletions (7% to 10% of cases). Cases with 13q deletions have the most favorable prognosis, whereas those with del(11q) or del(17p) have an unfavorable prognosis.185 The t(11;14) involving the cyclin D1 (CCDN1) gene has been described, but many of these cases are believed to be leukemic variants of mantle cell lymphoma. Deep sequencing studies of CLL have revealed a number of recurrent mutations, the most common of which involve the NOTCH1, MYD88, and SF3B1 genes.186
Clinical Presentation
Most patients with SLL present with painless generalized lymphadenopathy, which has frequently been present for several years. B symptoms are rare. Hepatosplenomegaly is present in less than 50% of patients. The peripheral blood in patients with SLL may be normal or reveal only a mild lymphocytosis; by definition, patients with SLL have an absolute lymphocyte count of <5,000/μL at the time of diagnosis. A serum paraprotein is found in about 20% of cases, and hypogammaglobulinemia is present in about 40%. Both CLL and SLL patients may develop autoimmune hemolytic anemia, pure red cell aplasia, and autoimmune thrombocytopenia. Elevated serum LDH is uncommon, whereas increased levels of serum beta-2 microglobulin are more frequently seen and can be a marker disease burden. SLL/CLL can transform to DLBCL (Richter syndrome), an event that is associated with a short survival.187 These patients present with rapidly growing masses, elevated serum LDH, and B symptoms. Rarely, transformation can be to B-cell prolymphocytic leukemia (B-PLL), which is characterized by high white cell counts and splenomegaly. It also has a poor prognosis.
Treatment of Small Lymphocytic Lymphoma
Patients with stage I SLL should be treated with involved field radiation, and not combined modality therapy or chemotherapy alone. In one limited series of 14 patients with stage I or II disease treated with 40 to 44 Gy, the 10-year freedom from relapse rates were 80% and 62% for stage I and stage II disease, respectively. Generally, patients with stage II or more advanced SLL are treated with chemotherapy regimens used for CLL (see Chapter 110). For patients with advanced stage disease who do not need systemic therapy but have one site causing symptoms, low-dose radiation (200 cGy for two fractions) can provide reasonable palliation, although the local control rates are not as high as seen with FL.145
Lymphoplasmacytic Lymphoma
Lymphoplasmacytic lymphoma represents about 1% of all NHLs. In some cases, patients present with mixed cryoglobulinemia, possibly related to concurrent hepatitis C virus infection.188,189
Pathology
Lymphoplasmacytic lymphoma is an indolent lymphoma composed of a diffuse proliferation comprised of a mixture of small lymphocytes, lymphoplasmacytic cells, and plasma cells.190 Immunoglobulin inclusions in the cytoplasm (Russell bodies) or invaginating into the nucleus (Dutcher bodies) are commonly seen. Unlike multiple myeloma, amyloidosis is rare. Occasional cases may also contain frequent larger immunoblast-like cells.
Immunophenotype and Genetics
Monoclonal cytoplasmic immunoglobulin is seen within the plasmacytoid cells and plasma cells by immunohistochemistry. The admixed lymphoid cells express B-cell antigens CD19, CD20, and surface IgM, and in general, do not express CD10 or CD23. A minor subset of cases is positive for CD5. Waldenström macroglobulinemia is an entity caused by high levels of monoclonal IgM that is generally associated with lymphoplasmacytic lymphoma (LPL). Deletions of 6q21 have been identified in 40% to 60% of patients with Waldenström macroglobulinemia. Activating mutations in MYD88, an adaptor protein that appears to function in signaling pathways downstream of the Ig receptor that lead to activation of the transcription factor nuclear factor kappa B (NF-κB) activation, are highly associated with LPL, being present in close to 100% of cases. However, mutations of MYD88 are not specific for LPL, because they are also seen less commonly in DLBCL and other low-grade B-cell NHLs.
Clinical Presentation
Clinically, this disease is similar to small lymphocytic lymphoma. The median age is early 60s, and nearly all patients have stage IV disease by virtue of bone marrow involvement. B symptoms and elevated serum LDH are uncommon. Lymph node and splenic involvement are common. In the WHO clinical study, 5-year OS (58%) and failure-free survival (25%) were similar to SLL.
Treatment
At least 25% of patients with LPL/waldenstrom’s macroglobulinemia (WM) have no indications for therapy at initial presentation. The indications for treatment include constitutional symptoms, cytopenias, or less commonly, symptomatic lymphadenopathy or splenomegaly. Other reasons for treatment are hyperviscosity related to the elevated serum IgM and paraneoplastic neuropathy.
Analogous to other indolent B-cell NHLs, rituximab plays a significant role in the therapy of LPL. Single-agent rituximab is indicated for minimally symptomatic patients. Approximately half of patients will have a partial response to single-agent rituximab.191 One can see transient increases in serum IgM levels after rituximab that can cause or exacerbate hyperviscosity.
Chemoimmunotherapy has largely replaced single agents for the treatment of LPL. Commonly used regimens include: dexamethasone, rituximab, cyclophosphamide (DRC)192; bortezomib plus rituximab with or without dexamethasone (BRD)193; or thalidomide plus rituximab.194 The latter two have limitations due to neuropathy. For DRC, the overall and complete response rates were 83% and 7%, respectively, and 2-year OS and PFS rates were 81% and 67%, respectively. Bortezomib and rituximab and thalidomide-rituximab have similar response rates. Alkylating agents, including chlorambucil and bendamustine, have RRs in excess of 80%. Purine analogs are active agents; however, stem cell toxicity can be an issue with purine analogs as well as chlorambucil.195 For recurrent disease, one can often utilize agents that were previously used. For patients with more refractory LPL, the mammalian target of rapamycin (mTOR) inhibitor everolimus, anti-CD52 mAb, and the oral Bruton’s tyrosine-kinase inhibitor, ibrutinib, are active. Selected patients with relapsed disease are considered for high-dose therapy with ASCT or alloSCT. The results seen are similar to that of other indolent lymphomas.
There are rare patients who have stage IE disease with this histology (i.e., renal involvement). In this case, modest dose RT (12 to 18 Gy) within the organ tolerance can provide long-term control and, occasionally, a cure.
Marginal Zone Lymphomas
MZLs are indolent NHLs that include three diseases arising from post-GC marginal zone B cells: splenic marginal zone B-cell lymphoma (± villous lymphocytes); extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT) type (MALT-type lymphoma, or MALT lymphoma); and nodal marginal zone B-cell lymphoma.196,197
Nodal MZL
Nodal MZLs constitute less than 1% of all NHLs. These lymphomas are primarily nodal diseases without evidence of extranodal involvement.
Pathology
Within lymph nodes, there are collections of B cells in a parafollicular, perivascular, and perisinusoidal distribution that often bear a monocytoid appearance, having folded nuclear contours and moderate abundant pale cytoplasm. These cells may surround reactive-appearing GCs and mantle zones. A subset of cases is also associated with variable degrees of plasmacytoid differentiation.
Immunophenotype and Genetics
Cells express monoclonal surface immunoglobulin (IgM > IgG > IgA) as well as CD19, CD20, CD79a, and are negative for CD10 and CD23. A minor subset of cases may be CD5 positive. Cases with plasmacytoid differentiation may show monoclonal expression of cytoplasmic kappa or lambda light chain by immunohistochemistry. Such cases may be associated with small monoclonal immunoglobulin spikes, but these are generally under 0.5 g/dL and are not associated with hyperviscosity. A subset of cases expresses surface IgD, analogous to splenic MZL. Immunoglobulin genes are rearranged with evidence of somatic mutation, implying a post-GC origin. There are no known chromosomal abnormalities specific to nodal MZL.
Clinical Features
Over 70% of patients present with stage III/IV disease, and the majority are asymptomatic. Bone marrow involvement is less common (45%) than in most indolent lymphomas. The 5-year survival for patients with nodal MZL is 55% to 79%. Similar to other indolent lymphomas, histologic transformation can occur with nodal MZL.
Treatment
The optimal therapy for patients with nodal MZL is not known. Patients are frequently treated with chemoimmunotherapy, typically either alkylating agents or purine analogs plus rituximab, which produce RRs in excess of 80%. A recent phase III study comparing CHOP-R to BR included 67 patients with MZL not otherwise specified.106 There was no difference (p < 0.32) in median PFS between CHOP-R (47 months) and BR (57 months). For now, patients should be offered either clinical trials or treated with regimens used for FL.
Splenic Marginal Zone Lymphoma (± Villous Lymphocytes)
Splenic MZL (± villous lymphocytes) constitutes less than 1% of all NHLs, with a median age of 65 to 70 years and uncommon before the age of 50 years.63 It is more common in Caucasians, with no gender predominance. Splenic MZL has been associated with viral infections, specifically hepatitis C and Kaposi’s sarcoma–associated herpesvirus (KSHV). In one study, treatment of hepatitis C induced regression of the lymphoma.
Pathology
In splenic MZL, there is an expansion of marginal zones in the spleen. Plasma cell differentiation may be seen in a subset of cases, but as in nodal MZL, monoclonal spikes, if present, are less than 0.5 mg/dL. Bone marrow, lymph nodes, and peripheral blood involvement (referred to as splenic lymphoma with villous or nonvillous lymphocytes) can also be present. Generally, cells have small nuclei, but in the peripheral blood, they typically have abundant cytoplasm with “shaggy” or villous projections.
Immunophenotype and Genetics
Splenic MZL cells express monoclonal surface IgM, IgD, CD19, and CD20. The tumor cells generally lack CD5 and CD10, helping to distinguish this tumor from SLL/CLL, MCL, and FL. They also typically are negative for CD25, CD103, and annexin A1, which helps to distinguish splenic MZL from hairy cell leukemia. Ig genes have evidence for somatic hypermutation in about half the cases. In splenic MZL, trisomy 3 is present in 39% of cases, which is found in other MZLs. Abnormalities of chromosome 7q are also frequently seen. Deep sequencing identified recurrent somatic mutations in genes involved in the NOTCH, NF-κB, and B-cell receptor pathways, as well as mutations in TP53.198 NOTCH2 mutations have been reported in 21% to 25% of cases, and were associated with a poor prognosis.
Clinical Features
Patients typically present with splenomegaly, lymphocytosis, and cytopenias, with lymphadenopathy being a much less common feature. B symptoms and elevated LDH are uncommon. Because of marrow and peripheral blood involvement, over 90% of cases have stage IV disease at diagnosis. IgM monoclonal gammopathies and mixed cryoglobulinemia can be seen, especially with a hepatitis C infection. Acquired C1 esterase deficiency seen in many B-cell lymphoproliferative disorders and can be a feature of splenic MZL.199 The survival of patients is in excess of 70% at 10 years. A prognostic model based on three risk factors—hemoglobin less than 12 g/dL, LDH level greater than normal, and albumin level less than 3.5 g/dL—could identify patients with 5-year cause-specific survivals of 88% for patients with zero risk factors, 73% for patients with one factor, and 50% for patients with two or three factors.200
Therapy
Similar to other indolent NHLs, many patients with splenic MZL do not require immediate therapy. Asymptomatic patients without splenomegaly or cytopenias can be observed. Patients with symptomatic splenomegaly and or significant cytopenias merit treatment. Those uncommon patients who also have hepatitis C may benefit from treatment of the infection, suggesting that tumor growth and survival is promoted by factors or signals elaborated in response to hepatitis C antigens. Splenectomy is reasonable for selected patients with excellent relief of symptoms and cytopenias. Splenectomy was associated with an overall response rate of 85% and an estimated PFS and OS at 5 years of 58% and 77%, respectively. For patients who are not surgical candidates, splenic radiation has some utility. In general, 150 cGy is given to the entire spleen three times per week. The total dose must remain under renal tolerance because the left kidney is almost always in the field. Single-agent rituximab can improve splenomegaly and cytopenias in over 90% of patients.201 In a study of induction with weekly rituximab followed by maintenance, the RR was 95%, with OS and PFS at 5 years of 92% and 73%, respectively.202 Other options for therapy at relapse are similar to those used for FL, and include retreatment with rituximab, alkylating agents, and purine analogs in combination with rituximab.
Extranodal Marginal Zone Lymphoma
MALT lymphoma is a subtype of MZL involving extranodal tissues. The most common site is the stomach, but MALT lymphoma has been described in a number of different organs and tissues including the skin, salivary glands, the lung, the small bowel, ocular adnexa, the breasts, the bladder, the thyroid, the dura, and the synovium. It has been associated with a variety of chronic inflammatory and infectious conditions, including autoimmune diseases such as Sjögren’s syndrome and Hashimoto’s thyroiditis, and infections with Helicobacter pylori (H. pylori), Borrelia burgdorferi (B. burgdorferi), Chlamydophila psittaci (C. psittaci), Campylobacter jejuni (C. jejuni), and hepatitis C virus (HCV).203–206 MALT lymphoma behaves indolently and is principally observed until symptoms related to organ impairment become evident; however, in many cases, early stage disease treatment with radiation therapy or antibiotic therapy appears to be curative. There are few dedicated studies of MALT lymphoma outside of early stage disease and much of the management of advanced stage disease is extrapolated from the FL literature, which often includes a small number of MZL patients.
Epidemiology
MALT lymphomas account for approximately 5% to 8% of all NHLs, but represent 50% to 70% of all MZLs.63,207 It is the third most common subtype of NHL after DLBCL and FL. The median age at diagnosis is 60 years, with incidence nearly equal in men and women. Two-thirds of patients present with stage I/II disease, with a minority of patients having more advanced disease at diagnosis. B symptoms and bone marrow involvement are rare. MALT lymphomas can transform into a more aggressive lymphoma, but this occurs rarely. The most common transformation is into an activated B-cell–like DLBCL.208 Nearly half of all MALT lymphomas involve the gastric mucosa, where over 60% are associated with an H. pylori infection.
Pathology
MALT lymphomas are malignancies of antigen-stimulated B cells, which normally reside in lymph nodes within the marginal zone that is found outside the mantle zones of B-cell follicles.197 Histologically, they are characterized by a monoclonal infiltrate of small- to medium-sized cells with abundant cytoplasm and irregular nuclear contours. Variable numbers of larger centroblast-like cells may also be present, and a subset of cases exhibit plasmacytic differentiation. An essential pathologic feature is the presence of lymphoepithelial lesions created by the invasion of mucosal glands and crypts by aggregates of lymphoma cells, producing an appearance that resembles the lymphocyte M-cell structures found in normal Peyer patches.
Immunophenotype and Genetics
MALT lymphomas are surface Ig positive, and are also positive for B-cell markers (CD19, CD20, CD79a, and CD22), and negative for CD5, CD10, CD23, and cyclin D1.44 Uncommonly, MALT lymphomas are CD5 positive and this is associated with a worse prognosis; these lymphomas may have cytogenetic changes such as trisomy 3 and del7q.209 Distinguishing MALT lymphomas from benign reactive lymphoid infiltrates may be difficult; in this circumstance, light chain restriction by flow cytometry or immunoglobulin heavy chain gene rearrangement studies by PCR can be helpful.
Other cytogenetic abnormalities that have been reported in MALT lymphomas include t(11;18), t(14;18), t(1;14), t(3;14), and trisomy 8. The t(11;18) is the most common; it occurs in 18% to 53% of MALT lymphomas of any site and is associated with a low-grade histology.210,211 It produces the fusion of the apoptosis inhibitor 2 (API2) gene and the MALT1 gene. The resulting fusion gene encodes a chimeric protein that stimulates the activation of NF-κB, a transcription factor that turns on a number of genes that promote proliferation and inhibit apoptosis.212 The t(11;18) translocation predicts for a poor response to H. pylori–directed therapies in gastric MALT lymphoma.213 A substantial proportion of malignant B cells in MALT lymphomas express B-cell receptors with strong homology to rheumatoid factors, and this appears to be mutually exclusive with the presence of the t(11;18) translocation.214 This suggests that t(11;18)-negative MALT lymphomas are driven by the stimulation of high-affinity B-cell receptors by antibody–antigen immune complexes and activated T cells, whereas t(11;18)-positive MALT lymphomas are not dependent on B-cell receptor signaling, but instead are driven by constitutive activation of NF-κB. The t(14;18), which pairs the MALT1 gene with the IgH gene and drives overexpression of MALT1 protein, is more common in nongastric MALT lymphomas.215 The t(1;14), which results in the overexpression of BCL10, is rarer overall but more frequent in gastric and pulmonary MALT lymphomas. The t(3;14) translocation is present in 10% of thyroid, ocular adnexal, and cutaneous MALT lymphomas.216 This translocation involves the IgH and FOXP1 genes and drives overexpression of the FOXP1 transcription factor. Of note, overexpression of MALT1, BCL10, and FOXP1 are all believed to result in NF-κB hyperactivation, making this a common feature of genetically diverse MALT lymphomas. MALT lymphomas are also more often associated with gains at chromosomes 3p, 6p, and 18p, and del(6q23) than the other subtypes of MZL.217
Clinical Presentation
The clinical presentation of MALT lymphoma depends in large part on the site of disease. Gastric and intestinal MALT lymphomas may present with symptoms of dyspepsia and abdominal pain, sometimes with signs and symptoms of bowel obstruction, but rarely with bleeding. These lymphomas are diagnosed on endoscopy with biopsies from multiple areas of endoscopically abnormal tissue as well as random sampling of macroscopically uninvolved mucosa. Involvement of the salivary and lacrimal glands, on the other hand, can result in Sjögren’s-like syndromes of dry eyes and mouth. MALT lymphomas involving the ocular adnexa typically present with painless conjunctival injection and photophobia, resembling allergic conjunctivitis. Patients with bronchus-associated lymphoid tissue (BALT) lymphomas typically are older men and can have symptoms including cough, fever, and/or weight loss.218 Other sites of disease often present with an obstructing mass. Some patients are diagnosed incidentally, either because of imaging studies or an exam of the eye or GI track done for another reason or as part of an evaluation for a monoclonal gammopathy, which is present in approximately 25% to 35% of MALT lymphoma patients; this feature is generally associated with plasmacytoid differentiation.219 B symptoms are rare in this disease.63 Bone marrow involvement is present in a minority of patients; therefore, cytopenias are rare, as is disease in the peripheral blood.
In addition to the blood tests that are standard for patients with NHL at diagnosis, patients with MALT lymphomas should have a few additional tests. HCV testing should be performed given its association with MALT lymphoma; an HIV virus test is advised. Additional laboratory studies to consider include a β2-microglobulin, serum protein electrophoresis and immunofixation, and serum light chains. Staging is done with CT scans of the chest, abdomen, and pelvis, as well as imaging of the neck, including the parotids and salivary glands, and orbits with CT or MRI. A bone marrow biopsy should be considered for patients with multifocal disease, and an evaluation of the gastric mucosa is reasonable for all patients with nongastric MALT lymphoma given the documented high rate of gastric involvement in these patients.220
Treatment
Management of MALT lymphoma depends both on stage and site of disease. As an indolent lymphoma with a long OS, close observation at diagnosis until the development of signs, symptoms, or organ function impairment as a result of the disease is appropriate for patients with advanced stage disease. An exception is patients with advanced stage MALT lymphoma and concomitant HCV infection; a trial of anti-HCV antiviral therapy in these patients may result in regression of their lymphoma. For patients with early stage and localized disease, however, treatment with radiation therapy or treatment with antibiotics, such as for H. pylori–positive gastric MALT lymphoma, has been associated with high RRs and durable responses, many of which may represent cures. Treatment of symptomatic or organ impairing relapsed, refractory, or advanced stage disease is similar to approaches used in FL with chemotherapy, immunotherapy, or chemoimmunotherapy.
Gastric MALT lymphoma represents a paradigm for treating early stage, localized MALT lymphomas. For those associated with an H. pylori infection that do not harbor a t(11;18) translocation, eradication of H. pylori is effective treatment and results in good long-term disease control and OS.221–223 In patients with H. pylori–negative lymphomas, MALT lymphomas with a t(11;18) translocation, or lymphomas that fail to respond to H. pylori therapy, RT is the preferred treatment modality.224 Chemotherapy, immunotherapy, or chemoimmunotherapy is active in this disease but is generally reserved for patients with relapsed or refractory disease to antibiotic therapy or RT, or patients with more advanced stage or aggressive disease.225,226 Similarly, MALT lymphoma of the ocular adnexa is primarily treated with RT.227 However, given the association described by some groups between C. psittaci infection and MALT lymphoma in this area, antibiotic therapy with doxycycline has been studied.228 The RR of single-agent doxycycline for MALT lymphoma of the ocular adnexa was 83%, with two-thirds of patients having partial responses. The 2-year PFS was 55%.
For relapsed or refractory disease or disease that is more extensive at presentation, agents that have been used and reported include single-agent therapy with alkylating agents such as chlorambucil or cyclophosphamide, purine analogs such as cladribine, bortezomib, and rituximab, and occasionally, multiagent anthracycline-based chemotherapy for younger patients with more aggressive disease. The use of single-agent, continuous, low-dose oral chlorambucil or cyclophosphamide in patients with early or advanced stage disease yielded CR rates of 75% and a relapse rate of 21% during the 8 year follow-up.229 Single-agent rituximab in patients with stage I through IV MALT lymphoma (15 gastric, 10 nongastric) who were either chemotherapy naïve or who had progressed following chemotherapy resulted in an overall response rate of 73% and was better for chemotherapy-naïve patients than for previously treated patients (87% versus 45%).225 Duration of response was short, however, with 36% of responders progressing at a median of 10.5 months. Combination chemoimmunotherapy with rituximab and fludarabine results in response rates of 85% to 100% and 2- to 3-year PFS of 80% to 100% at the expense, however, of significantly greater toxicity.230 MALT lymphoma is extremely sensitive to radiation therapy, and this modality has a role in the palliation of patients with advanced disease.
Mantle Cell Lymphoma
MCL is a malignancy of monomorphous small- to medium-sized B cells with the characteristic t(11;14) leading to overexpression of the cyclin D1 cell cycle regulator in the majority of cases.
Pathology
MCLs are neoplastic counterparts of naïve “mantle zone” B cells. Morphologically, MCL can have either diffuse architecture, or a vaguely nodular appearance, occasionally growing predominantly in expanded mantle zones around reactive GCs. Cytologically, in most cases, the neoplastic cells are small- to medium-sized and have irregular nuclei and scant cytoplasm. Some cases of MCL have a predominance of intermediate-size cells with more open “blastic” chromatin; such blastic variants are associated with a high mitotic rate. Other cases are comprised of a spectrum of cells, including large cells (pleomorphic variant).
Immunophenotype and Genetics
MCLs express B-cell antigens, surface IgM (usually together with surface IgD), CD5, and CD43 and usually lack CD10 and CD23. Overexpression of cyclin D1 further distinguishes these tumors from most other entities. IgH variable gene segments lack a somatic mutation in 84% of cases (pre-GC), with the remainder being mutated.231 By FISH, greater than 90% of MCLs have the t(11;14) associated with the rearrangement of the cyclin D1 gene (CCDN1). The remaining cases do not overexpress cyclin D1, but instead usually overexpress cyclin D2, cyclin D3, or cyclin E due to the presence of translocations involving these genes and the IgH locus.232 Cyclin D1–negative cases are similar clinically to cyclin D1–positive cases233 and have a similar gene expression profile. Deep sequencing234 has identified NOTCH1 mutations in a minority of cases, which may be associated with a poor prognosis. SOX11 overexpression is also associated with a worse prognosis.232,235
Clinical Features. MCL constitutes about 7% of all NHLs. About 75% of patients are males, with a median age of 63 years. Approximately 70% of patients have stage IV disease, and B symptoms are observed in approximately one-third of patients. Typical sites of involvement are the lymph nodes, the spleen, the liver, Waldeyer’s ring, and bone marrow. Peripheral blood involvement is present in 25% to 50% of patients at presentation. MCL can involve any region of the GI tract (88% lower tract, 43% upper tract by endoscopy), occasionally presenting as multiple intestinal polyposis.236 CNS involvement is rare and is usually associated with a leukemic phase.
The median survival of patients with MCL is 4 to 5 years and improving. Approximately 10% to 15% of patients have a disease with a more indolent disease, with minimal lymphadenopathy, mild splenomegaly, and a proliferation index measured by Ki-67 staining of around 10%.237 These patients have a disease that behaves more like an indolent NHL, where a watchful waiting approach does not compromise response to therapy or survival. In contrast, patients with the blastic variant at diagnosis have a median survival of 18 months. Blastic transformation occurs in 35% of patients, with a risk of 42% at 4 years, and once occurring, the median survival is 3.8 months.238
Prognostic models have been employed for patients with MCL. The IPI developed for diffuse aggressive NHLs provides stratification of patients. Attempts to improve on the IPI include the mantle cell lymphoma International Prognostic Index (MIPI), which includes age, performance status (PS), LDH, and white blood cell count (WBC)58 as prognostic factors, and several reports show that the MIPI is better than the IPI at stratifying patients.239 The proliferation index alone and also when incorporated into the MIPI provides additional predictive power.240 Gene expression profiling has been examined in MCL patients. In those studies, the proliferation signature and high expression of cyclin D1 were associated with an unfavorable prognosis.241 Mutations and deletions of p53 are also associated with a worse prognosis.242
Treatment. The majority of patients with MCL have a disseminated disease requiring treatment. An indolent behaving disease is seen in 10% to 15% of patients, where a delay of initiation of treatment was not deleterious.241 The treatment of MCL historically involved single alkylating agents as well as combination chemotherapy (CVP, CHOP) to which 30% to 50% of patients had a CR, with a median duration of 1 to 3 years.243 In a meta-analysis, single alkylating agents offered results similar to combination chemotherapy.244 A small number of patients with MCL will present with stage I to II disease. These patients are potentially curable with combined chemotherapy and involved field radiation (30 Gy). Chemoimmunotherapy has shown a significant impact in the treatment of MCL. A meta analysis of 638 patients with MCL showed that rituximab-containing regimens significantly increased median survival (37 versus 27 months)245 as compared to chemotherapy alone. One of the mainstays of chemoimmunotherapy for the initial treatment of MCL is CHOP-R. However, results of a randomized trial comparing CHOP-R to BR reported superior PFS with BR with less toxicity.106 In patients with a median age of 70 years, the median PFS for BR was 35 months, compared to 22 months with CHOP-R. One other randomized study compared CHOP-R to the fludarabine, cyclophosphamide, rituximab (FCR) regimen. In that study, for patients over age 60 years, CHOP-R and FCR had similar CR rates, but CHOP-R had less toxicity, and the OS at 4 years was 62% versus 47% in favor of CHOP-R.246 This study included a second randomization of maintenance with interferon-α or rituximab until progression. For the patients who received CHOP-R, a significant survival benefit from maintenance rituximab was observed, with an estimated 4-year OS rate of 87% versus 63%.
More aggressive approaches for the initial treatment of MCL have been the rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone (R-HyperCVAD) regimen or the consolidation of first remission following chemoimmunotherapy with high-dose therapy and ASCT. The most recent update from the M.D. Anderson Cancer Center of R-HyperCVAD reported a median OS not reached at 8 years, and a median time to failure of 4.6 years.247 A multi-institution SWOG phase 2 trial of R-HyperCVAD reported median PFS and OS of 4.8 and 6.8 years, respectively. However, 39% of patients were unable to complete the planned treatment.248
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


