Non-Hodgkin Lymphomas
Non-Hodgkin lymphomas (NHL) are a heterogeneous group of malignancies of the lymphoid system characterized by an abnormal clonal proliferation of B cells, T cells, or both. Scientific knowledge regarding NHL has increased dramatically in the past two decades, resulting in specific advances in the spheres of molecular biology and immunobiology and leading to new histopathologic classifications and therapies.
 EPIDEMIOLOGY
EPIDEMIOLOGY
The U.S. age-adjusted incidence rate for NHL was 19.8 per 100,000 between 2004 and 2008, according to the Surveillance, Epidemiology, and End Results (SEER) program of the National Cancer Institute.1 In 2010, the estimated number of new NHL cases in the United States was 65,500; deaths from NHL were 21,000.2 In 2008, worldwide estimated incidence was 356,000 new cases and 191,000 deaths. International NHL incidence rates vary, with the highest incidence rates in North America, Europe, and Australia/New Zealand. The lowest rates have been reported in Asia and the Caribbean.3 NHL is primarily a disease of older populations, with a median age of 65 at diagnosis.4
There has been a striking increase in NHL incidence rates over the past four decades, with a doubling between 1970 and 1990. The rate of increase has slowed since 1990, with a modest increase in incidence from 18.5 per 100,000 to 20.5 per 100,000 between 1990 and 2005.5 Incidence rates have remained at approximately 20 to 21 per 100,000 over the past decade. The mortality rate for NHL has risen steadily, peaking in the late 1990s, with more recent decline over the past decade. The U.S. mortality rate rose from 5.6 per 100,000 to 8.2 per 100,000 between 1975 and 2000. Since a peak of 8.9 per 100,000 in 1997, the mortality rate has steadily declined with most recent SEER data reports of 6.5 per 100,000 deaths in 2007.6 Similar increases have been noted in international cancer registries.7
The increased incidence of NHL has been partly attributed to advances in molecular diagnostic techniques, aging of the population, immunosuppression from human immunodeficiency virus (HIV), infectious agents, and occupational or environmental exposures.8 There are likely other contributing factors that remain unknown at present.
 ETIOLOGY
ETIOLOGY
Several genetic diseases, environmental agents, and infectious agents have been associated with the development of lymphoma. Familial clustering of NHL has been described. However, it is not clear whether the familial aggregations are due to hereditary factors or shared environmental exposures.9
Immunodeficiency
The frequency of NHL is greatly increased in immunocompromised patients. The two most common clinical circumstances are among HIV-infected patients and solid organ transplant recipients, both associated with prolonged immunosuppression.10,11 In HIV patients, NHL is the second most common malignancy, with a rate of 1.2% per year.12 The introduction of highly active antiretroviral therapy (HAART), however, has resulted in a decline with subsequent stabilization in the incidence. Treatment outcomes of NHL in this population have improved as well.13,14
Patients with autoimmune and chronic inflammatory disorders, including Sjögren syndrome, Hashimoto’s thyroiditis, systemic lupus erythematosus, and less commonly, celiac sprue also have an increased risk of NHL.15,16 Several rare inherited disorders are associated with up to a 25% lifetime risk for development of lymphoma.17 These include severe combined immunodeficiency, hypogammaglobulinemia, common variable immunodeficiency, Wiskott-Aldrich syndrome, Chediak-Higashi syndrome, and ataxia-telangiectasia. Lymphomas associated with these disorders are often Epstein-Barr virus (EBV) related and usually highly aggressive in their behavior.
Infectious Agents
A number of viral infectious agents are implicated in the pathogenesis of NHL. EBV is associated with Burkitt’s lymphoma, posttransplant lymphoproliferative disorders (PTLD), acquired immunodeficiency syndrome (AIDS)-associated primary central nervous system lymphoma (PCNSL), congenital immunodeficiency associated lymphomas, and natural killer (NK) T-cell lymphomas.18 The human T-cell lymphotropic virus type 1 (HTLV-1) is an RNA virus responsible for adult T-cell leukemia/lymphoma (ATL).19 In endemic areas, more than 50% of all NHL cases are ATL, although the risk for development of disease is only approximately 5% in infected patients. Human herpes virus 8 (HHV-8), the causative agent for Kaposi’s sarcoma, is also associated with several rare lymphoproliferative diseases, including primary effusion lymphoma.20 The hepatitis C virus (HCV) is linked with several NHL subtypes, including splenic marginal zone lymphoma.21,22
The strongest association between infectious agents and NHL is in marginal zone lymphomas (MZL). The bacterium Helicobacter pylori has been linked to gastric mucosa-associated lymphoid tissue (MALT) lymphomas.23,24 It has been suggested that several other bacteria, including Borrelia burgdorferi, Campylobacter jejuni, and Chlamydia psittaci, may also play a role in the pathogenesis of MZL of other sites.21,25,26
Environmental and Occupational Exposures
Occupations associated with a higher risk of developing NHL include farmers, teachers, dry cleaners, butchers, printers, wood workers, mechanics, and agricultural workers.27,28 Several studies have shown an increased risk of NHL in relation to pesticide exposure, particularly phenoxyl herbicides and organochlorines.29,30 The development of NHL has also been linked to hair dyes, organic solvents, high levels of nitrates in drinking water, arsenic, pesticides, fungicides, lead, vinyl chloride, and asbestos.29 Radiation has been suggested as a causative agent with increased lymphoma incidence in survivors of nuclear explosions or atomic reactor accidents.31 NHL is also observed as a late effect of prior radiation therapy or chemotherapy.32 Dietary factors and tobacco and alcohol use may affect the risk of developing NHL.33
TABLE 78.1 WORLD HEALTH ORGANIZATION 2008 CLASSIFICATION OF MATURE


 PATHOLOGY AND IMMUNOBIOLOGY
PATHOLOGY AND IMMUNOBIOLOGY
NHL is a group of many different disease entities, often difficult to diagnosis, with a correspondingly complex histopathologic classification that has changed relatively frequently over the years. The changing classifications reflect new knowledge gained as well as difficulties with older systems that were recognized with the passage of time, such as interobserver variability, difficulties with reproducibility, and a somewhat confusing picture with respect to clinical–pathologic correlates. The predecessor to the currently utilized World Health Organization (WHO) classification was the Working Formulation.34 It subdivided NHL by biologic behavior; low grade, intermediate grade, and high grade; and indolent or aggressive behavior groups. This staging characterization has been abandoned in the new WHO classification. Such groupings were convenient and appeared clinically useful, but represented an oversimplification and did not account for several distinct clinical–pathologic entities.
The WHO 2008 classification divides NHL into B-cell and T-cell neoplasms, with over 40 different lymphomas delineated, as shown in Table 78.1.35 These specific entities are distinguished on the basis of reproducibly identifiable morphologic, immunologic, and genetic characteristics. The specific diseases described may be either indolent or aggressive in behavior. Within a given disease there may be a range of behaviors (e.g., anaplastic large cell lymphoma [ALCL]). Similarly, the histologic grade and the biologic behavior may vary within a specific disease entity (e.g., follicular lymphoma [FL]). In addition, the WHO classification describes new disease categories not clearly recognized in the Working Formulation, notably marginal zone lymphoma (MZL), mantle cell lymphoma (MCL), peripheral T-cell lymphomas (PTCL), ALCL, and primary mediastinal large B-cell lymphoma.
A description of all the varieties of NHL is not practical and beyond the scope of this chapter. This chapter will focus on the most common entities in order of decreasing frequency, namely, diffuse large B-cell lymphoma (DLBCL), FL, MZL, MCL, PTCL, and chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL), although other less common entities are also discussed. Incidence data are derived largely from the NHL Classification Project,36 which evaluated 1,403 cases of NHL at nine study sites around the world, establishing the frequency of the subtypes, geographic variation in incidence, and clinical correlates.
It must be emphasized that lymphoma pathology is complex with a long history of interobserver disagreement. National Comprehensive Cancer Network (NCCN) guidelines describe as “essential” specialized hematopathology review of all slides and adequate immunophenotyping to establish the diagnosis.37 Fine-needle aspiration alone is rarely acceptable for the initial diagnosis of lymphoma due to lack of nodal architecture.38
Diffuse Large B-Cell Lymphoma
DLBCL is a neoplasm of large, transformed B cells with a diffuse growth pattern and a high (>40%) proliferation fraction. The cells may resemble centroblasts, immunoblasts, multilobated cells, or anaplastic large cells. It is the most common type of NHL (31% of all cases, 33% if primary DLBCL of the mediastinum is included).36 DLBCL is a heterogeneous group of neoplasms with multiple distinct variants described in the current WHO classification.35 In addition, DNA microarrays (oligonucleotides or cDNA probes) show that three different subgroups of DLBCL with unique molecular abnormalities can be identified.39 These include the germinal center B-cell subtype and the activated B-cell subtype. Primary mediastinal DLBCL also has a unique molecular signature that closely resembles classical Hodgkin lymphoma (HL).40 Models predictive of clinical outcome using gene-expression data have also been developed.41–42,43 Treatment tailored to molecular subclassification of this disease is premature at present but may become part of future routine clinical practice.
DLBCL express one or more pan B-cell markers (CD19, CD20, CD22, and CD79a), as well as CD45, and often surface immunoglobulin.44 Twenty-five percent to 80% in various studies express BCL-2 protein.45 Approximately 70% express BCL-6 protein, consistent with a germinal center origin.45,46 Most cases of DLBCL have somatic mutations in the immunoglobulin variable region genes, suggesting they have progressed through the germinal center where immunoglobulin affinity maturation occurs. The BCL-2 gene is rearranged in 15% to 30% of cases, the C-MYC gene is rearranged in 5% to 15%, and the BCL-6 gene is rearranged in 20% to 40% of cases.45
Follicular Lymphoma
The next most common type of NHL is FL (22% of all cases). In the Working Formulation, it was described as low grade or indolent. It has also been referred to as follicle center cell lymphoma.36 In North America, the frequency is somewhat higher at 31% versus 14% at other geographic sites.47 Thus, in North America FL and DLBCL are approximately equal in frequency.
FL is a tumor of follicle center B cells (centrocytes and centroblasts) with a follicular (nodular) pattern that morphologically is similar to normal germinal centers.48 The neoplastic follicles may be present in the entire tumor, or the lymphoma may contain a diffuse component as well. FL is graded based on morphology. There is either a predominance of small cleaved cells (grade 1), a mixture of small cleaved and large cells (grade 2), or predominantly large cells (grade 3). In the WHO classification, the number of large cells per high-power field (0 to 5, 5 to 15, >15) is used to assign grades (1 to 3, respectively). However, if there are diffuse areas comprised predominantly of blastic cells, a diagnosis of DLBCL is also made.
Clinically, grades 1 and 2 are closely related with no apparent differences in biologic behavior or response to therapy. FL grade 3 tends to have a somewhat higher relapse rate with an outlook favorably influenced by anthracycline-containing chemotherapy. Although grade 3 FL is not the same as DLBCL, it may contain areas of the latter, which further suggests the need for more aggressive therapy.
The tumor cells of FL are usually surface immunoglobulin positive, express pan-B-cell–associated antigens (CD19, CD20), CD21, CD10 (60% of the time), but lack CD5. Most cases are BCL-2 positive; nuclear BCL-6 is expressed by at least some of the neoplastic cells. T(14;18) and BCL-2 gene rearrangement are present in the majority of the cases (85%). BCL-2 protein is expressed in most cases, ranging from 100% in grade 1 to 75% in grade 3 FL.49
Marginal Zone Lymphomas
MZL is now recognized as a distinctive subtype of NHL in the WHO classification, accounting for approximately 10% of all cases of NHL. MZL entities include nodal MZL, splenic MZL, and extranodal MZL of MALT. MALT as a distinct clinical pathologic type of lymphoma was first described in 1983.50
Extranodal MZL is characterized by a polymorphous infiltrate of small lymphocytes, marginal zone (centrocyte-like) B cells, monocytoid B cells, and plasma cells, as well as rare large basophilic blast cells (centroblast- or immunoblast-like). In epithelial tissues, the marginal zone B cells typically infiltrate the epithelium, forming lymphoepithelial lesions defined by invasion and partial obstruction of mucosal glands by tumor cells.51 Although transformed large cells are typically present, they are in the minority. If present in large numbers, a diagnosis of DLBCL is warranted.
The tumor cells express surface immunoglobulin and lack immunoglobulin D. Forty percent to 60% have monotypic cytoplasmic immunoglobulin, indicating plasmacytoid differentiation. They express pan B-cell–associated antigens (CD19, CD20, CD22, CD79a) but are generally negative for CD5 and CD10. There is no specific marker for MZL at present. Immunophenotyping studies are useful in confirming malignancy (light chain restriction) and in excluding B-cell chronic lymphocytic leukemia (B-CLL; CD5+), mantle cell (CD5+), and follicular lymphomas (CD10+).52
Immunoglobulin genes are rearranged; the variable region has a high degree of somatic mutation, as well as intraclonal diversity consistent with a postgerminal center stage of B-cell development.53 The most common reported cytogenetic abnormalities are trisomy 3, seen in 60%, and t(11;18), seen in 25% to 40% of patients.52,54 These changes are characteristically found in extranodal but not nodal MZL.
The most common site of MALT is the stomach. Lymphoid tissue is not normally present in the stomach, but in response to an antigenic stimulus brought about by H. pylori, normally present T cells in the gastric mucosa attract a B-cell population, giving rise to lymphoid follicles and, after prolonged antigenic stimulation, lymphomas.55
Peripheral T-Cell Lymphomas
PTCLs are the group most confusing to clinicians. They were not a separate entity in the Working Formulation but were frequently classified as either diffuse poorly differentiated lymphocytic lymphoma or diffuse mixed lymphocytic-histiocytic lymphoma. The term peripheral T-cell lymphoma is often misinterpreted. It refers not to the anatomic distribution of the lymphomas but to their origin from so-called peripheral or mature T cells outside the thymus, as opposed to thymic (precursor) T lymphocytes. The T-cell lymphomas collectively make up approximately 10% of all NHL.56 They are a diverse group that includes 14 different entities (Table 78.1).
PTCLs constitute the most frequently occurring variety of T-cell lymphoma. In the International Lymphoma Study Group (ILSG) report, PTCLs comprised 7% of the total cases, making them approximately equal in frequency to MZL, SLL, and MCL.56 Their frequency is quite different, however, by geographic locale. ILSG data showed a roughly 3% incidence of PTCL in North America compared with 9% in South Africa, Hong Kong, and London.47 EBV may be associated with T-cell lymphomas originating in the nasal cavity.57
PTCLs typically contain a mixture of small and large atypical cells. The architectural pattern is diffuse. T-cell–associated antigens (CD2, CD3, CD4) are variably expressed, with some tumors expressing CD8. Sometimes the T-cell antigens CD5 and CD7 are lost. B-cell–associated antigens are lacking. The T-cell receptor genes (TCR) are usually, but not always, rearranged. No specific cytogenetic or oncogene abnormality has been reported.
ALCL is a special variant of PTCL.58,59 In the ILSG project, ALCL comprised 2% of all NHL.36 The tumor is usually composed of large cells with round, pleomorphic, or horseshoe-shaped nuclei with single or multiple prominent nucleoli and abundant cytoplasm, giving the cells an epithelial or histiocyte-like appearance. The cells express CD30 (Ki-1) and usually express CD25 and either T-cell or null lineage–specific antigens.58 CD30 was originally recognized on HL cells. In some cases, there may be confusion between ALCL and HL, but distinction between the two on the basis of immunophenotyping and morphology is usually possible.
The overexpression of a novel tyrosine kinase gene on chromosome 2 known as anaplastic lymphoma kinase (ALK) is a characteristic feature of ALCL.59 Approximately 60% of cases overexpress the ALK protein; such cases have a better prognosis than ALK-negative cases, except for skin cases. ALCL in children or young adults is usually ALK positive.59
Small Lymphocytic Lymphoma
SLL is the nodal equivalent of B-CLL.48 In the WHO classification these are considered a single entity (CLL/SLL). It is a neoplasm composed predominantly of small lymphocytes with condensed chromatin and round nuclei. Larger lymphoid cells (prolymphocytes and paraimmunoblasts) with more prominent nucleoli and dispersed chromatin are always present, usually clustered in pseudofollicles. SLL comprises approximately 7% of all NHL.
The tumor cells of SLL express human leukocyte antigen (HLA)-DR, B-cell–associated antigens (CD19, CD20, CD22, CD79a), and both CD5 and CD23 and have faint surface immunoglobulin. CD23 is particularly useful in distinguishing CLL/SLL from MCL.
Approximately 50% of cases have abnormal karyotypes.60 Trisomy 12 is reported in one-third of cases with cytogenetic abnormalities and correlates with atypical histology and an aggressive clinical course.60 Abnormalities of 13q are reported in no more than 25% of the cases and are associated with long survival. CLL/SLL can transform to DLBCL (Richter syndrome).61
Mantle Cell Lymphoma
MCL was first distinguished in the 1980s by Weisenburger et al.,62 who described a type of FL in which there were wide mantles of malignant cells around apparently benign germinal centers. The term mantle zone lymphoma was proposed, subsequently modified to mantle cell. It was thought to represent a variant of FL and was classified under the Working Formulation as a low-grade lymphoma. Its behavior, however, is more characteristic of aggressive disease. MCL represents about 7% of all NHL.36
Since the original description by Weisenburger et al.,62 additional features of this disease have been recognized. It is a neoplasm of small to medium-sized B cells with irregular nuclei that resemble the cleaved cells (centrocytes) of germinal centers. The morphologic pattern may be diffuse, nodular, a mantle zone, or some combination thereof. Tumor cells are typically CD5 positive, CD23 negative, CD20 positive, and CD10 negative. A characteristic cytogenetic abnormality is t(11;14) involving the BCL-1 gene and resulting in the overexpression of cyclin D1. The product of the cyclin D1 gene can be detected in paraffin-embedded tissue sections with the immunoperoxidase technique and is useful in distinguishing MCL from other lymphoma variants.63,64
 CLINICAL FEATURES: GENERAL
CLINICAL FEATURES: GENERAL
Nodal Versus Extranodal Disease
NHL is primarily a disease of older adults (in contrast to HL), with a median age at presentation from 55 to 65 years.65–67 There is a slight male over female preponderance (55% to 60% male). NHL may involve lymph nodes in almost any area of the body but may also present in extranodal sites, presumably arising from lymphoid tissue widely distributed throughout the body. Approximately two-thirds of NHL is nodal at presentation and one-third extranodal, again in contrast to HL, where extranodal presentation is rare.65,68
Patients with primarily nodal disease usually present with an asymptomatic lump in the neck or inguinal area; B symptoms (fevers, night sweats, weight loss) may be present in 20% to 30% of patients. There is often a history of some spontaneous regression and then regrowth of the nodes.
The most frequent sites of nodal involvement are the neck in approximately 70% of patients, the groin in approximately 60%, and the axilla in approximately half.66,67 Although patients with nodal presentations may appear to have localized disease initially, full staging evaluation results in assignment to a more advanced stage in two-thirds.69
Patients presenting with extranodal lymphoma, on the other hand, usually have localized disease. Indeed, some authorities conclude that the definition of primary extranodal disease should be restricted to those with stage I or II disease.70 The symptoms relate to the site of involvement. The gastrointestinal (GI) tract is the most common (25% to 35% of extranodal disease), followed closely by Waldeyer’s ring and other head and neck sites (18% to 28%) and skin.68,71 Epigastric discomfort, abdominal pain or bleeding, and sore throat or difficulty swallowing are among the usual symptoms for these locations.
The histologic picture of most primary extranodal lymphomas is that of DLBCL, although depending on anatomic site, other histologies are commonly seen (e.g., MALT lymphoma of the stomach). Nodal presentations, conversely, are more commonly FL. Nodal and extranodal disease of similar histologic type, stage, and other prognostic variables are treated in an equivalent fashion and have a similar outcome.72,73 For example, DLBCL, stage IA in the neck, treated with combination chemotherapy and radiotherapy, would have the same outcome as stage IA DLBCL in the stomach treated similarly. These characteristics are summarized in Table 78.2. There are notable exceptions, however. For example, localized disease in the central nervous system (CNS) has a much worse prognosis as does primary testicular disease and perhaps lymphoma in the breast (to be discussed subsequently).74,75,76
TABLE 78.2 CHARACTERISTICS OF NODAL VERSUS EXTRANODAL NON-HODGKIN LYMPHOMA
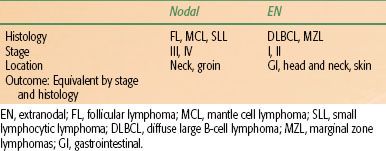
TABLE 78.3 CLINICAL, LABORATORY, AND RADIOLOGIC EVALUATION OF PATIENTS WITH NON-HODGKIN LYMPHOMA

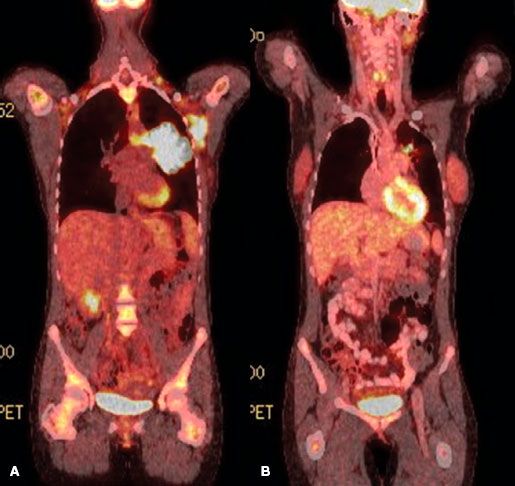
FIGURE 78.1. Fused coronal positron emission tomography (PET) computed tomography images of a patient with diffuse large B-cell lymphoma before (A) and after (B) chemotherapy. The postchemotherapy PET was scored as positive in the mediastinum. Biopsy confirmed persistent disease.
Evaluation and Staging
A careful history and physical examination are required. The history should record presence or absence of B symptoms (fever, night sweats, weight loss), performance status, duration of lymph node enlargement and growth history, and any specific symptoms suggestive of extranodal involvement. All peripheral nodal areas should be examined clinically, enlarged nodes measured with suitable calipers and two-dimensional measurements recorded, and inspection of the skin, oral cavity, and tonsils performed. Patients with suspected or established head and neck lymphoma or GI tract involvement should undergo direct fiberoptic laryngoscopy or endoscopy.77 Staging procedures are summarized in Table 78.3.
The use of functional imaging, initially with gallium, now largely with positron emission tomography (PET) using 18F-fluorodeoxyglucose (FDG) has increased dramatically in the past decade.78,79,80,81,82 PET has largely replaced gallium because of greater accuracy, particularly in the abdomen, convenience, and the ability to perform at one examination an integrated PET and computed tomography (CT) scan.79,83 The fused images obtainable from an integrated PET-CT increase the accuracy of diagnosis by more precisely localizing anatomically areas of increased isotope uptake (Fig. 78.1). The fused images are also very helpful to the radiation oncologist in planning radiation fields. A number of authors in relatively small series have suggested that PET imaging is the most sensitive indicator of disease present initially, surpassing CT scans in this regard with a sensitivity exceeding 90%, compared with 60% to 70% for conventional imaging.80,81
Some authors have suggested PET imaging at diagnosis is unnecessary in the presence of obvious generalized disease and should be omitted to conserve resources and reduce costs.84 Because PET is so often now performed to evaluate response (however, see below), a pretreatment scan is quite useful for comparison if resources permit. Further, PET-CT scanning (contrast-enhanced CT) eliminates the need for a separate CT.
PET with gallium scanning is particularly useful after therapy, where residual anatomic abnormalities on CT scan are common.85,86,87–88 Positive functional imaging studies after therapy are a poor prognostic sign, predicting for early relapse and suggesting that minimal cell kill has been accomplished by the therapy, because enough viable cells remain to take up the isotope in question. In contrast, patients with residual anatomic abnormalities as imaged on CT who have negative PET or gallium studies enjoy an outlook comparable with PET negative or gallium-negative patients without residual static abnormalities. A positive PET scan following salvage chemotherapy for relapsed DLBCL (in preparation for high-dose chemotherapy) has also been reported as a poor prognostic sign and a relative contraindication to proceeding with transplant.89,90
Other important staging studies include bilateral bone marrow aspirate and biopsy and routine blood studies, particularly lactate dehydrogenase (LDH), because this is an important prognostic factor. Patients should be evaluated for the presence of hepatitis B antigen because of concerns of reactivation of this virus with chemotherapy and particularly if rituximab is administered.91
Other staging studies will be indicated for suspected primary extranodal disease depending on the site of involvement. For example, lymphoma arising in the head and neck region may require magnetic resonance imaging (MRI) for precise anatomic delineation or lumbar puncture for CNS evaluation in the case of testicular or sinus lymphoma (see specific lymphoma sections for additional details).
The most widely used staging system is the anatomically based Ann Arbor system (Table 78.4), originally devised at a conference in Rye, New York, in 196592 and modified at Ann Arbor, Michigan, in 197093 and again at the Cotswolds conference in England in 1988.94 The principal changes introduced at Cotswolds were the use of the subscript “x” to designate “bulky” disease (i.e., a mass of ≥10 cm in maximum diameter), and the definition of criteria for liver or spleen involvement as evidence of focal defects with two or more imaging modalities. Abnormal liver function study results were to be ignored for staging purposes.
The Cotswolds conference and a subsequent workshop95 also discussed categories of response to treatment.96 It has been long recognized that many patients had good clinical responses with improvement but not complete disappearance of disease on follow-up static imaging such as CT scans. Although the Cotswolds conference did not consider functional imaging in the assessment of response, it has been incorporated by the International Working Group.96 A complete response has now been redefined to include residual masses on CT scan that have become PET negative.
Prognostic Factors
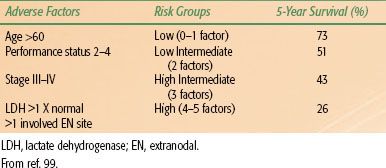
Anatomic stage of disease, tumor mass, and systemic symptoms are important prognostic indicators. Other prognostic variables investigated have included patient age, performance status, histologic type of lymphoma, tumor size, number of nodal or extranodal sites, tumor phenotype (B or T cell), LDH, β2-microglobulin levels, serum albumin, hemoglobin, and proliferation indices.97,98 Many of these variables were shown to be significant in univariate and multivariate analyses but in small series from individual institutions.
In an attempt to develop a better prognostic model, the International Non-Hodgkin Lymphoma Prognostic Factors project examined data on 2,031 patients, all with aggressive-histology NHL.99,100 Two indices were developed, the International Prognostic Index (IPI) and the Age Adjusted International Prognostic Index, because age was found to be a highly significant prognostic variable (>60 years vs. ≤60 years). For both indices, four risk groups were identified—low, low intermediate, high intermediate, and high—depending on the number of risk factors present in a given patient. Five prognostic variables were found to be significant: age, performance status, stage (I/II vs. III/IV), number of extranodal sites, and LDH (Table 78.5). When patients were divided by age, three factors remained independently significant: performance status, stage, and LDH.
TABLE 78.4 THE ANN ARBOR/COTSWOLDS STAGING CLASSIFICATION FOR HODGKIN LYMPHOMA AND NON-HODGKIN LYMPHOMA

TABLE 78.5 INTERNATIONAL PROGNOSTIC INDEX FOR DIFFUSE LARGE B-CELL LYMPHOMA
With the widespread utilization of rituximab therapy and improved outcome for patients with DLBCL, some authors have suggested revisions to the IPI to allow for more separation of prognostic groups.101 Essentially these authors regrouped the IPI into three rather than four groups, with the best outlook for those with zero adverse factors (94% survival), a good outcome for those with one or two adverse factors (80%), and a poor outcome (55% survival) for those with three or more adverse factors. The German High-Grade Non-Hodgkin Lymphoma Study Group has found, however, the IPI to still be a valid predictor in the rituximab era.102 Others have suggested an elderly prognostic index (for patients older than 70).103 At present, IPI remains the standard with no consensus on modifications.
The IPI was developed primarily for patients with DLBCL. It has also been successfully applied with some modifications to patients with PTCL.104,105,106 There were initial attempts to apply the IPI to FL, but here it was less useful because most patients fell into a favorable prognostic category. A recent international effort has addressed this problem, however.107 Patient characteristics were collected from 4,167 patients with FL diagnosed between 1985 and 1992. Five adverse prognostic factors were identified on multivariate analysis, including age (>60 years vs. <60 years), stage (I/II vs. III/IV), hemoglobin level (<120 g/L vs. ≥120 g/L), number of nodal areas (>4 vs. ≤4), and serum LDH. Three risk groups were identified: low risk (zero to one adverse factor), intermediate risk (two factors), and poor risk (three or more adverse factors). Patients were approximately equally divided among the three groups. There was good separation between the groups in terms of survival (Table 78.6). The resulting index is known as the follicular lymphoma international prognostic index (FLIPI).
The FLIPI has subsequently been revised to predict for progression-free survival (PFS) in the rituximab era.108 The significance of FLIPI-2 remains to be determined with additional follow-up studies. In the original FLIPI study, neither tumor size nor histologic grade was considered. Other groups have suggested these are important variables.109
The molecular features of DLBCL have been examined using gene-expression profiling as assessed by DNA microarrays or polymerase chain reaction (PCR). These studies have involved patients receiving chemotherapy for DLBCL who had been classified according to the IPI risk categories.43 One study used 13 key genes, another 17 to divide patients into two or four groups, respectively, with markedly different survival rates, accounting for IPI risk categories.42,43 In general, the outcome for patients with germinal center B-cell–like DLBCL differs from that of activated B-cell–like DLBCL, the latter being considerably worse.108
Gene-expression profiling has also been investigated in FL,110,111 with two reports differing significantly, in that one examined the molecular features of tumor infiltrating cells and the other the actual lymphoma cells. Dave et al.110 were able to group the patients into four quartiles with widely disparate median lengths of survival depending on two gene-expression signatures. This trial controlled for clinical features as determined by the IPI but not by the FLIPI. Similarly, the study by Glas et al.111 used gene-expression profiling to predicate clinical behavior. Again, the IPI was the clinical classification utilized rather than the FLIPI. Further investigation is needed to determine the value of gene-expression profiling in FL if patients are subdivided according to the FLIPI rather than the IPI. Additionally, it remains to be determined whether the tumor-infiltrating immune cells are the appropriate targets for gene-expression profiling or the tumor itself. In general, gene-expression profiling is not widely available clinically and must still be considered investigational.
A number of other biomarkers have been investigated for their influence on outcome in DLBCL, FL, and other histologic variants of lymphoma. Results, however, have been inconsistent so that clinical indices remain the standard prognostic evaluation tool at this time.45,112
TABLE 78.6 FOLLICULAR LYMPHOMA INTERNATIONAL PROGNOSTIC INDEX

 CLINICAL–HISTOPATHOLOGIC CORRELATES
CLINICAL–HISTOPATHOLOGIC CORRELATES
The pathology and immunobiology of the most frequently encountered varieties of NHL have already been discussed. This section examines the clinical features of the most commonly encountered NHLs. Data from the NHL classification project are invaluable in this regard.36,113
Diffuse Large B-Cell Lymphoma
Patients most often present with an enlarging peripheral nodal mass or with symptoms related to a primary extranodal site of involvement, such as abdominal or epigastric pain. DLBCL is primarily a disease of older adults, with a median age of 64 years and a slight preponderance of men (55%). B symptoms are present in approximately one-third of patients. Just over half the patients (55%) have localized disease (stages I or II) at onset. Just over half the patients with stage I or II disease have extranodal presentations. When subdivided according to IPI score, one-third of patients have a score of 0 or 1, one-half a score of 2 or 3, and the remainder a score of 4 or 5.36,113 Lymphomas that present extranodally are most commonly DLBCL.72
The WHO recognizes several distinct variants of DLBCL. The most notable include primary mediastinal DLBCL, T-cell/histiocyte-rich large B-cell lymphoma, and primary DLBCL of the CNS (discussed later).
Primary mediastinal DLBCL is believed to arise from thymic medullary B cells. Microarray studies have revealed a unique molecular signature for primary mediastinal DLBCL with a resemblance to nodular sclerosis HL.114 There are characteristic genetic changes, notably absence of BCL2 and BCL6 rearrangements, as well as consistent increases in chromosome 9P and 2P, the former being rather specific for primary mediastinal DLBCL and observed in up to 75% of cases.
Primary mediastinal DLBCL comprises about 2.4% of all NHL, about 7% of all DLBCL. The disease affects primarily young women (median age 37) and is generally confined to the anterior mediastinum, sometimes with supraclavicular or cervical adenopathy. Relapses tend to be extranodal, however. When grouped according to the IPI, the prognosis is similar to that of DLBCL generally, perhaps a bit more favorable,114,115 with a plateau observed in the PFS curve after 18 to 24 months. Rituximab appears to improve outcomes to the same extent as other subtypes of DLBCL.116 The role of consolidation radiation therapy (RT) has been questioned, given the more favorable prognosis. However, omitting consolidation RT for early-stage primary mediastinal DLBCL has not been formally studied.115
T-cell/histiocyte-rich B-cell lymphoma consists of large malignant B cells with a florid background of inflammatory T cells, with or without histiocytes. Clinically, it occurs in a younger population compared with DLBCL, generally with a male predominance.117,118 This entity is also more likely to present with B symptoms and involve the spleen, liver, and bone marrow. Treatment recommendations are similar to DLBCL, not otherwise specified.
Follicular Lymphoma
FL affects primarily older adults (median age, 59 years). There is a slight female preponderance. In contrast to DLBCL, approximately 70% of patients present with generalized disease, most with stage IV disease. The bone marrow is the principal extranodal organ involved. Localized extranodal presentation is uncommon, reported in only 6% of the ILSG series, again in contrast to DLBCL.113 FL, in general, has a favorable outcome in the intermediate term, with 5- to 8-year survival rates from 70% to 80%. The failure-free survival (FFS) rate is considerably less, however, at approximately 40%.119–121 Furthermore, there is little evidence of flattening of the survival curves with time, suggesting that relapse occurs continually over the course of many years. An exception may be the small percentage of patients who present with localized disease and are treated definitively with radiotherapy, a significant number of whom may be cured with that treatment.122,123 Despite being considered an indolent disease, the leading cause of death is lymphoma, often after histologic transformation to DLBCL.
Marginal Zone Lymphoma
MZL includes both nodal and extranodal varieties and the unique entity of splenic MZL.124 The more familiar name for extranodal MZL is MALT lymphoma. Approximately 70% of MZL is extranodal, with the remainder divided about equally between splenic and nodal MZL. Two-thirds to three-fourths of patients have stage I or II disease, the former more common than the latter. The prognosis is generally excellent with 85% to 90% 5-year survival irrespective of stage, although FFS is less.74,125–127 In the MD Anderson Cancer Center series, splenic MZL had the best survival (93% at 5 years), although the disease was invariably stage IV at diagnosis, confirming the indolent nature of these diseases.128
Extranodal MALT lymphoma occurs primarily in the stomach, but a number of other anatomic sites are commonly seen, including the thyroid, parotid glands, orbit, and skin. Recently, an association with bacterial infection for MALT at sites other than the stomach has been described.21 C. psittaci has been found in many cases of ocular adnexal lymphoma; antibiotic treatment has been shown to result in lymphoma regression in some series but not in others.129,130 Additionally, B. burgdorferi and C. jejuni have been associated with MZL arising in the skin and small intestine.21,131 Unlike gastric MALT, the standard initial treatment for these conditions, however, remains local radiotherapy, with very high rates of complete response and local control in excess of 90%, similar to what has been reported for gastric MALT.127,132
Splenic MZL patients usually present with splenomegaly.126,133,134 Almost all have stage IV disease, principally because of bone marrow involvement. The disease is relatively indolent, with three-quarters of patients alive at 5 years, but a more aggressive subset does exist. The most effective therapy appears to be splenectomy. Indeed, the diagnosis is usually not clearly established until this time. Radiotherapy to the spleen has infrequently been used.
Peripheral T-Cell Lymphoma
The International Peripheral T-Cell Lymphoma Project recognized 12 varieties of PTCL, accounting for 5% to 10% of NHL in Western countries but 5% to 20% of NHL in Asia where NK/T-cell lymphoma and adult T-cell leukemia/lymphoma are far more common.135,136 This section will discuss only the more common variants.
Most common in the West is PTCL, not otherwise specified (NOS), accounting for about one-third of cases (vs. 20% in Asia). This is a disease of older adults, with a median age of 61 years.56,113,137–139 Slightly more than half of the affected patients are male. Most patients present with nodal disease in a similar fashion to B-cell lymphoma. In contrast to DLBCL, however, the great majority of patients (70%) have stage III or IV disease at diagnosis.137,139 Approximately half of the patients have B symptomatology. The IPI also tends to be more advanced, with 52% of patients having a score of 2 or 3 and 31% with a score of 4 or 5. Many patients have some preceding disorder of the immune system (27% in the U.S. combined series) such as angioimmunoblastic lymphadenopathy, mononucleosis, lymphomatoid granulomatosis, or papulosis.137
Extranodal NK/T-cell lymphoma is a T-cell lymphoma of special interest. This lymphoma has a variety of names in the older literature, including angiocentric lymphoma, midline malignant reticulosis, polymorphic reticulosis, and lethal midline granuloma.140–143 It is much more frequent in Asia (about 22% of T-cell lymphomas) than in the United States and is often associated with EBV.144 In contrast to most lymphomas, tissue destruction of the nasal or facial area is common. The response to chemotherapy and radiotherapy is variable and slow, in contrast to the usual rapid response observed in most other lymphomas.145
ALCL is another T-cell lymphoma. Two forms may be distinguished: a systemic illness with widespread involvement of lymph nodes and extranodal sites and a type primarily limited to the skin.59,146,147–150 The systemic type may in turn be separated into those patients who are ALK positive and those who do not overexpress this protein. The clinical features and prognosis differ widely among these two categories. Patients with ALK-positive ALCL are predominantly young men and, despite advanced-stage disease, respond well to combination chemotherapy, with survival rates in the range of 75% to 90%. Patients who are ALK negative, in contrast, tend to be older with a more nearly equal male to female ratio. Their response to chemotherapy is much worse, with survival rates reported in the 20% range.
Cutaneous ALCL constitutes a special situation.146 It is almost invariably ALK negative but carries an excellent prognosis. It is often difficult to distinguish from benign lymphomatoid papulosis (LyP). The latter may spontaneously remit and tends to run a benign clinical course over many years. ALCL of the skin is quite responsive to localized radiotherapy (see the section on lymphomas of the skin).151
Overall ALCL has one of the best survival rates of any lymphoma, approximately 75% at 5 years in the ILSG study, despite its aggressive appearance under the microscope. The heterogeneity and complexity of this particular variant of NHL well illustrates past difficulties of attempting to group lymphomas into categories of low grade, intermediate grade, and high grade, indolent or aggressive, favorable or unfavorable, on the basis of histologic appearance alone.
Small Lymphocytic Lymphoma
SLL is morphologically and immunotypically identical to CLL; the two are classified as one entity by the WHO. Of combined cases, about 85% are CLL and 15% SLL. Clinically, SLL is distinguished from CLL by the absence of peripheral blood involvement and <30% infiltration of the bone marrow. SLL is generally manifest by widespread nodal involvement with or without hepatosplenomegaly.152 In those uncommon instances where it is localized, however, radiotherapy may make it curable. The median age is 65 years, the oldest for any lymphoma. The male to female ratio is approximately even. Ninety percent of cases are generalized (i.e., stages III or IV), with 90% of those being stage IV (or 80% of the total). This is truly an indolent lymphoma. Survival may be prolonged even in the absence of therapy. In the ILSG project, the overall survival (OS) rate was approximately 50% at 5 years; the FFS rate was considerably less, however, at 25%.
Mantle Cell Lymphoma
MCL has a 74% male preponderance and a median age of 63 years.113,153 Eighty percent of patients have stage III or IV disease at onset. IPI scores are high, with 23% of patients with a score of 4 or 5, 54% with a score of 2 or 3, and only 23% with a score of 0 or 1. In common with FL, the organ most likely to be involved is the bone marrow, which is positive in two-thirds of patients at the time of diagnosis. GI tract involvement is frequent as well.
Although originally classified among the low-grade/indolent tumors in the Rappaport system, the clinical course for MCL is unfavorable in the great majority of patients. The 5-year survival rate in the ILSG project was only 27%, with an FFS rate of 11%.113 This FFS rate is, in fact, among the worst for virtually any type of lymphoma. The poor overall 5-year survival rate is matched only by PTCL and lymphoblastic lymphoma. The shape of the survival curve is continually negative, with no plateau to suggest cure in any significant percentage of patients. The situation is somewhat analogous to FL, but the latter usually has a much more prolonged natural history.
 PRINCIPLES OF TREATMENT
PRINCIPLES OF TREATMENT
Surgery
Before modern radiotherapy and chemotherapy, surgical resection constituted the only potentially curative treatment for NHL. It was commonly used for extranodal sites such as the stomach or head and neck and was curative in a significant percentage of patients when the disease was truly localized. Similarly, patients with nodal presentations and localized disease were managed by radical surgical procedures. The surgical approach subsided rapidly with the development of radiotherapy and chemotherapy and the recognition of the unique radiosensitivity and chemotherapy responsiveness of lymphomas.
Surgery, however, is still widely used to establish a diagnosis by biopsy. That may involve a major surgical operation, such as exploratory laparotomy for the diagnosis of stomach, intestinal, retroperitoneal, or mesenteric lymphoma. This is becoming much less common, however, with the use of endoscopic and laparoscopic techniques. With an established diagnosis of lymphoma, surgical resection as primary treatment should be a rare event, with radiotherapy and chemotherapy forming the mainstays of treatment. For example, lymphoma of the stomach should almost never be primarily resected, as the results are equal or better with radiotherapy with or without chemotherapy.
Radiation Therapy
Dose of Radiation Therapy When Used Alone
Malignant lymphomas are, in general, uniquely sensitive to ionizing radiation. For the great majority of anatomic locations, the sensitivity of the tumor is greater than that of the corresponding normal tissue, usually by a considerable amount, a luxury not available when treating most solid tumors. As a consequence, radiation fields can often be somewhat larger to cover potential microscopic areas of spread. Three-dimensional treatment planning and intensity-modulated RT may be less important for lymphomas than for solid tumors because of the lower doses generally used but have a definite role in many situations.
Dose–response data for RT of NHL are sparse. Most data come from phase II retrospective analyses; almost all of these analyses were carried out years ago, well before the latest WHO pathologic classification. The diseases most often studied were what we now know as DLBCL and FL, with a reasonable amount of information also available for MALT lymphomas. On the other hand, diseases such as PTCL, MCL, and ALCL have never been analyzed separately, so one is forced to extrapolate from the data for DLBCL and FL.
The classic articles in this regard are from Stanford University and Princess Margaret Hospital. Fuks and Kaplan154 in 1973 reported that doses in the range of 44 Gy achieved local control of FL in >95% of instances. For diffuse histiocytic lymphoma (corresponding roughly to DLBCL), local failure rates, however, were in the range of 20% to 30%, regardless of the dose of RT delivered (Fig. 78.1). These data have been widely misinterpreted as suggesting or justifying a dose of 50 Gy for DLBCL. In fact, they suggest a subset of resistant disease in the range of 20%, regardless of the dose of RT delivered.
A series of articles from Princess Margaret Hospital also addressed this issue.155–157 Dose–response curves were constructed for both diffuse histiocytic lymphoma (mostly DLBCL) and FL. For DLBCL patients with medium- or large-bulk disease, defined as 2.5 to 5 cm in size and >5 cm, respectively, an approximately 50% local control rate was achieved with a dose of 20 Gy, rising to 70% at 30 Gy and 80% at 40 Gy with a plateau thereafter, and no apparent improvement with additional dose (Fig. 78.2). For patients with small volume (<2.5 cm) DLBCL, a local control rate >90% was achieved regardless of dose. For patients with nodular (follicular) disease, doses in the range of 25 to 35 Gy produced a local control rate >90%.158
Similar data were reported in a more contemporary series from the University of Florida.159 For patients with low-grade lymphomas treated with RT alone (mostly FL), doses of 30 Gy achieved local control in >90% of patients. For those with intermediate- or high-grade disease, doses of 30 to 50 Gy also achieved local control in >95% of instances.
There was a suggestion that tumor bulk appeared to influence the outcome. Patients with tumor size >6 cm were treated with combined modality therapy (CMT). It was suggested that doses of at least 40 Gy were necessary in these circumstances for optimal local control, although the data demonstrate local failure in 1 of 51 patients treated with >40 Gy as part of a CMT program versus 4 of 70 patients treated with doses of 30 to 40 Gy.
For MZL, particularly MALT of the stomach, high local control rates are achieved with doses of approximately 30 Gy. Data from Princess Margaret Hospital demonstrate a 96% complete response (CR) with 4% partial response (PR) and an overall local control rate of 95%.132,160,161 In a smaller series of patients from Memorial Sloan-Kettering Cancer Center, the response and local control rates were 100% with similar doses (30 Gy).162 One hundred percent local control with 30 Gy was also achieved in a Harvard series.127 Similar data, although in smaller numbers, are available for MZL at sites other than the stomach.
It is unclear if tumor size affects the dose required for local control in FL and MZL. Smaller tumors may do well with <30 Gy.
Recently a phase III dose–response trial was carried out by the British National Lymphoma Investigation (BNLI) group.163 This multicenter trial randomized 1,001 patients between 1997 and 2005, 361 with indolent lymphomas, predominantly FL and to a lesser extent MZL, and 640 with aggressive lymphomas, predominantly DLBCL, between two different radiation schedules: 24 Gy versus 40 to 45 Gy for indolent disease and 30 Gy versus 40 to 45 Gy for aggressive disease. About 20% of indolent disease patients had received chemotherapy, but 80% of aggressive histology patients did. Thus, this is primarily a trial of the dose of RT alone for indolent disease and the dose of RT in a combined modality program for aggressive disease. Tumor bulk was not considered in the randomization process.
For indolent disease the outcomes (survival, PFS, and local control) were virtually identical regardless of dose. The latter approximated 70% at 10 years (Fig. 78.2). From these data and the phase II data cited above, there is no justification for RT doses exceeding 30 Gy for indolent disease when RT is used as a single modality.
Dose of Radiation Therapy in a Combined Modality Therapy Program
For DLCBL, almost all patients, including those with localized disease, are treated with CMT, now typically R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy followed by RT. Thus, a more relevant question than the dose of RT required for local control in patients treated with RT alone is the required dose in a CMT program.
There are many phase II reports in the literature of CMT for DLBCL with rather widely varying doses of RT used. The Vancouver group reported 308 patients with stage I and II DLBCL treated with CHOP (and related combinations) followed by involved field radiotherapy (IFRT) to doses of 30 to 35 Gy (2 to 3 Gy per fraction).164 The 10-year cause-specific survival rate was 82%. In-field local failures occurred in 3% of patients.
Investigators at the MD Anderson Cancer Center reviewed 469 patients with DLBCL treated between 2001 and 2007 with R-CHOP (6 to 8 cycles) with or without RT. Forty percent had stage I or II disease and 60% had stage III or IV. Overall, 30% had consolidation IFRT following CR to chemotherapy with doses of 30 to 39 Gy. Local control was achieved in 100% of patients with all relapses outside the RT field.165
Krol et al.166 at the Daniel den Hoed Cancer Center in Rotterdam looked at 26 versus 40 Gy for patients with stage I DLBCL who had experienced a CR to CHOP. There was no difference in outcome for the two doses in this retrospective analysis.
At Duke University, the authors examined 45 patients with stage I and II DLBCL treated with CHOP who experienced a CR, defined by anatomic imaging and the presence of a negative gallium scan at the completion of therapy. Doses of RT ranged from 10 to 50 Gy but were clustered largely around 30 Gy. Durable local control was achieved in 92% of patients.
The phase III data from the BNLI again demonstrate virtually superimposable curves for local control, PFS, and OS for 30 versus 40 to 45 Gy.163 Again, there is no mention of bulk disease in the BNLI study nor was functional imaging commonly employed to determine CR.
FIGURE 78.2. Freedom from local progression (A), progression-free survival (B), and overall survival (C) for indolent and aggressive non-Hodgkin lymphomas according to radiation dose. (From Lowry L, Smith P, Qian W, et al. Reduced dose radiotherapy for local control in non-Hodgkin lymphoma: a randomised phase III trial. Radiol Oncol 2011;100:86–92, with permission.)
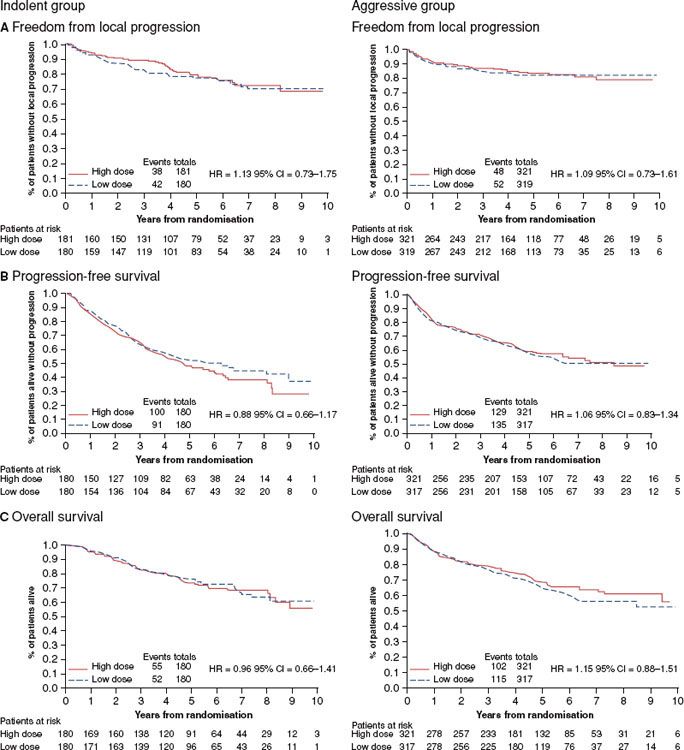
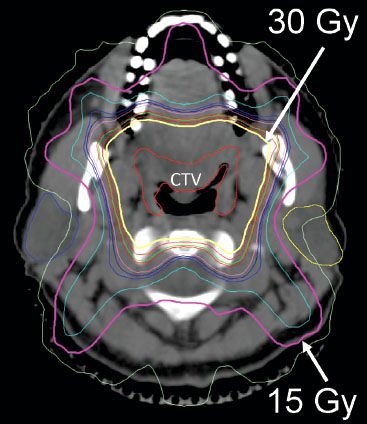
FIGURE 78.3. Axial image from planning computer tomography scan of patients with marginal zone lymphoma of Waldeyer’s ring (base of tongue and bilateral tonsils). The clinical target volume (CTV) is labeled. The planning target volume was a 1-cm expansion around the CTV. The 15 Gy (pink) and 30 Gy (yellow) isodose lines are marked with white arrows. Note sparing of the parotid glands.
The issue of the dose of RT for FL or MZL in CMT programs has usually not been considered because of the lack of efficacy of chemotherapy for localized FL or MZL. Thus, the optimal RT dose in CMT programs (for aggressive histology patients achieving CR with chemotherapy) appears to be 30 Gy, on the basis of the phase II and III data cited above. Even smaller doses may suffice and are under investigation at Duke University. There is no convincing evidence that so-called bulk disease requires more, particularly if PET negative after systemic therapy. For patients who respond to chemotherapy but have persistent PET-positive disease (Fig. 78.1), higher doses of consolidation RT may be required to achieve optimal local control (~40 Gy).167
An unresolved and controversial question is what to do with the patient who is PET positive after chemotherapy. This section will address only the dose of RT to be used if RT alone is selected for treatment. The BNLI study found no difference in outcome between 30 and 40 to 45 Gy for the small number of aggressive histology patients treated with RT without chemotherapy, but no details are given. Nonetheless, in view of the phase II data above, the authors favor a dose 40 Gy under these circumstances.
Field Size and Treatment Volume
The optimal treatment volume or field size for RT of localized NHL is also a matter of some controversy, because definitive phase III trials to resolve the issues are not available. Many of the conclusions regarding appropriate field size are extrapolated from information regarding patterns of failure.
For DLBCL, the pattern of failure after CMT is usually disseminated disease, with a small percentage with local failure.168 After chemotherapy alone, more local failure occurs.168,169 Failure in nodal areas adjacent to the original disease is uncommon.
The question frequently arises as to the appropriate treatment volume or field size when the patient with DLBCL has experienced CR to chemotherapy. Should one treat the original prechemotherapy tumor volume, the postchemotherapy tumor volume, or the original volume plus adjacent nodal areas? What kind of margin should be employed? The policy at Vancouver, for example, was to cover the nodal or extranodal area in question with a margin of about 5 cm, without, however, specifying whether to treat the prechemotherapy or postchemotherapy volume.164
In view of the patterns of failure data cited above, treatment of the original volume plus adjacent nodal areas has pretty much been abandoned. Thus, IFRT seems most appropriate. Whether the involved field is the pre- or postchemotherapy tumor volume depends a lot on where the original tumor was and the tolerance of surrounding normal tissues. With DLBCL of the stomach, for example, the entire organ would be treated regardless of the response to chemotherapy. For patients presenting with nodal disease in the neck unilaterally, the entire neck on that side would generally receive RT after chemotherapy. It would not be necessary or desirable to “prophylactically” treat Waldeyer’s ring for neck presentations of DLBCL.
For tonsil, base of tongue, or nasopharynx presentations of DLBCL, after a CR to chemotherapy, the specific primary site should be radiated with appropriate three-dimensional planning at a minimum or IMRT (Fig. 78.3). One would usually not irradiate all of Waldeyer’s ring but only the specific primary site. In the absence of clinical involvement of the neck at diagnosis, it would not be necessary to “prophylactically” treat the neck. Indeed, omission of neck radiation in these instances would facilitate parotid-sparing treatment plans. Mouth dryness seems to be a real problem for these patients, despite a relatively modest dose of radiation.
Conversely, for a large mediastinal mass, concerns about excessive pulmonary toxicity usually lead to treatment of the reduced tumor mass in the lateral dimensions or the normal mediastinal and hilar structures in the event of a CR to chemotherapy, not the original tumor volume.170
For FL, a number of authors have reported on patterns of failure and appropriate field sizes for patients with stage I or II disease.158,171 For patients presenting with nodal disease, most studies have suggested that FFS is improved with the use of total lymphoid irradiation (TLI), as opposed to IFRT.123,157,159,172 None of these studies has shown an improvement in OS, however, leading most centers to conclude that the morbidity and expense of TLI are not justified.170
Recently the Vancouver group has explored the use of involved nodal RT compared with IFRT, the former defined as covering just the involved nodal group with a margin of up to 5 cm and the latter defined as the involved nodal group and one or more immediately adjacent uninvolved nodal groups.109 This retrospective study was done in 237 FL patients. There were no significant outcome differences.
RT may be used alone to treat stage IIIA FL, although seldom done currently.173 In this instance, TLI would be required. The techniques of TLI (i.e., the mantle, para-aortic nodes, spleen, and pelvis) are discussed in Chapter 77.
Two additional specialized techniques may be applicable to NHL. Total body irradiation (TBI) was used years ago for palliation of advanced FL but has largely been supplanted in this regard by a variety of newer systemic agents. It is still used, however, as part of various regimens of high-dose chemotherapy (HDC) in conjunction with stem cell transplantation.174 When used in the setting of a myeloablative conditioning regimen, TBI is typically administered in a dose of 12 to 15 Gy, 1.2 to 2.0 Gy per fraction, once or twice daily, all depending on institutional preference. One randomized trial of single-dose (10 Gy) TBI (dose rate 0.125 Gy/min) versus 14.85 Gy in 11 fractions over 5 days (dose rate 0.25 Gy/min) revealed similar therapeutic efficacy but a lesser degree of veno-occlusive disease in the fractionated group.175 Both groups had the lungs shielded after 8 to 9 Gy. In the setting of a nonmyeloablative conditioning regimen, 2 Gy TBI is often utilized.
Patients are treated at an extended source to skin distance, the exact arrangement depending on the geometry of the treatment room. At Duke University, patients typically sit on a stretcher with the knees drawn up. Treatment is administered utilizing lateral fields. Unlike TBI for acute leukemia, a testicular boost is not used. The arms are crossed over the chest to provide some self-shielding of the lungs. Dose to the lungs is usually in the neighborhood of 8 to 10 Gy for a 13.5 Gy overall dose, to reduce the risk of pneumonitis. Most, but not all, institutions use some type of lung shielding to reduce the lung dose to approximately 8 to 10 Gy. Other toxicities of TBI include nausea, vomiting, diarrhea, and suppression of the blood counts. This dose would also be expected to cause permanent ovarian ablation in the majority of premenopausal women, sterility in men, and the long-term risk of cataract formation.176,177 In contrast, the acute and late toxicity of 2 Gy TBI in nonmyeloablative regimens are expected to be much less.
Whole-abdomen irradiation (WAI) may also be undertaken more frequently in NHL compared with HL. Mesenteric lymph nodes are commonly involved in NHL, unlike HL. If treating with CMT in a patient with stage II disease and widespread abdominal involvement, the radiation oncologist might wish to use WAI or a modified version excluding the pelvis or liver. The usual Duke University technique is opposed anterior and posterior fields from the diaphragm to the superior portion of the pelvis or the inferior portion of the obturator foramen with partial shielding of the iliac bones and femoral heads. Unless there is known liver involvement, the right lobe of the liver is also shielded. If doses <18 Gy are being used, kidney blocks are not used. If higher doses are desired, either kidney blocks need to be used or different field arrangements made. This depends on the exact location of the disease to be treated. If treating only mesenteric nodes, cross-table lateral fields may be appropriate. Three-dimensional planning is done in most patients. The kidneys are easily localized with CT simulation or with intravenous contrast on a conventional simulator. Modified WAI may also be undertaken as single-treatment modality for FL of the mesentery.
Radiation Therapy for Palliation
RT is a very effective palliative agent and should be considered more often in patients not responding to multiple courses of drug treatment.170 Often relatively small doses of radiation in brief courses can be quite effective. A Dutch trial described 304 sites treated in 109 patients with indolent lymphomas (mostly FL).178 Patients received 4 Gy, either in 1 or 2 fractions, to the symptomatic areas. The overall response rate was 92% with a CR in 61% of patients and a PR in 31%. A French trial had similar results.179 The median time to local progression was 25 months. In this study the number of prior chemotherapy regimens did not influence the response rate. Patients with FL that is behaving in an indolent fashion may sometimes be managed with judicious palliative irradiation for many years without systemic therapy.122,143 For critical local problems occurring in the palliative setting, such as spinal cord compression, RT is the treatment of choice. In this setting, where the goal is to maximize the chances of long-term freedom from local progression, doses of 30 Gy (2 Gy per day) would achieve that objective in 90% to 95% of instances.
For the more aggressive NHL histologies, responses to such low doses would not be predicted, but, in fact, response rates of 50% to 80% have been reported with a dose of 2 Gy é 2 and a median time to progression of about 1 year.180,181 Certainly effective palliation can be accomplished with doses well below 30 to 40 Gy.
Chemotherapy
Chemotherapy forms the mainstay of treatment for the great majority of patients with NHL, because these diseases are most often generalized. As with radiation, malignant lymphomas are, in general, very responsive to chemotherapy. This responsiveness, unfortunately, does not translate to cure of the patient in most instances. Of all the pathologic variants of NHL in the WHO classification, consistent curability with advanced disease is seen only in patients with DLBCL and to a lesser extent in some patients with PTCL and ALCL. Some of the more “indolent” lymphomas such as FL do not appear curable with conventional chemotherapy. A few patients with advanced indolent disease may be curable with an allogeneic transplant.
A large variety of drugs are available for the treatment of malignant lymphomas, including alkylating agents such as cyclophosphamide, corticosteroids, vinca alkaloids, purine analogs, and anthracyclines. They are typically used in combination in order to circumvent problems of drug resistance. The most widely used combination for the treatment of DLBCL has been CHOP.182 For patients with advanced DLBCL, this combination produces an approximate 50% to 60% CR rate, just over half of which are durable responses, for an overall cure rate of approximately 30% to 40%. Results are significantly improved by the addition of the anti-CD20 antibody rituximab, so that R-CHOP has rapidly become the new standard.183,184
Similarly, R-CHOP is probably the most widely used combination in the United States for the treatment of FL,185 although its superiority to other less aggressive combinations has not been established in phase III studies. These data are discussed in greater detail in the sections on the specific types of NHL.
Stem Cell Transplantation and High-Dose Chemotherapy
There has been a great interest in the application of HDC with stem cell rescue in the treatment of malignant lymphomas, both for relapsed disease following initial treatment and for those patients deemed to be at high risk for relapse at diagnosis. The underlying concept is that larger doses of conventional chemotherapy will result in greater tumor cell kill and increased cure rates. The doses involved are so large that they would be lethal because of hematopoietic toxicity without a rescue strategy. Accordingly, hematopoietic progenitor cells are harvested from the patient before the HDC, either from the bone marrow itself or more often mobilized from the patient’s peripheral blood and then reinfused to re-establish marrow function (autologous stem cell transplantation [ASCT]). The high frequency of bone marrow involvement in certain types of NHL limits this strategy, as well as chemotherapy resistance.
Alternatively, an allogeneic transplant may be carried out in individuals with a suitable matched donor in which the stem cells are harvested from the donor. In this procedure, it is hoped that the infused donor stem cells will additionally mount an immunologic attack on the tumor. Allogeneic transplantation may be preceded by full-dose (myeloablative) chemotherapy designed to have not only an antitumor effect, but also to condition the patient for the infusion of the donor cells, or it may be preceded by a nonmyeloablative or reduced intensity conditioning (RIC) program designed primarily to enable the recipient to accept the donor stem cells. In this latter situation the major antitumor effect is postulated to derive from the infused donor stem cells. RIC allogeneic transplants are associated with a much lower treatment-related mortality (10% to 20%) compared with myeloablative allogeneic transplants (40% to 50%).186,187 TBI is often a component of the conditioning program, with doses varying quite widely from 2 to 13.5 Gy (see the section Principles of Treatment: Radiation Therapy).
Studies of ASCT have been carried out in many varieties of NHL but primarily in DLBCL. Numerous phase I, II, and III trials have been reported. An expert committee of the American Society for Blood and Marrow Transplantation has recently comprehensively reviewed all published trials and issued recommendations.188
In general, ASCT has been investigated in three types of situations: (a) patients who have been treated with conventional chemotherapy and then relapsed; (b) patients who fail conventional chemotherapy from the onset (so-called primary refractory disease); and (c) patients who have responded well to primary chemotherapy but are considered at high risk for relapse. The most widely accepted use is for the treatment of patients with DLBCL who have relapsed following initial CHOP or R-CHOP chemotherapy. In a phase III trial from the Parma group, patients with DLBCL who had relapsed following initial CHOP chemotherapy and who were responsive to a salvage program (dexamethasone, cisplatin, cytarabine [DHAP]) were then randomly assigned to receive either four additional cycles of DHAP or a high-dose chemotherapy program.189 Those receiving the HDC program had a markedly improved FFS and OS compared with those getting conventional chemotherapy (46% FFS vs. 12%, 53% OS vs. 32%). Note that in both arms of this trial, IFRT to original bulky sites of disease (≥5 cm) was utilized, with a dose of 35 Gy in 20 fractions in the conventional chemotherapy arm and 26 Gy in 1.3 Gy fractions twice a day in the HDC arm. All patients in the Parma trial were <60 years of age. Patients with a favorable IPI score of 0 did not benefit.190 Those with a short remission after initial chemotherapy had a worse outcome.191
The Parma trial and associated phase II studies have led to the adaptation of ASCT as standard of care for patients <60 years of age with DLBCL relapsing after initial chemotherapy, although the Parma trial is the only phase III investigation of relapsed DLBCL patients ever done. It is also important to note that in this as well as almost all other trials, patients who do not respond to the initial salvage program do poorly with subsequent HDC and are not considered good candidates. PET scanning has also been utilized to define response; those with a persistently positive PET after a salvage program do poorly with HDC.89
A number of groups have explored the incorporation of HDC programs into initial therapy for patients with aggressive histology lymphomas considered at high risk for relapse. Between 1999 and 2010, two meta-analyses and nine prospective randomized trials have examined this issue and are described by the American Society for Blood and Marrow Transplantation expert committee. Rituximab was not included in any of these trials. The committee concluded the evidence was insufficient to recommend HDC/ASCT for any patient group.188
The final issue addressed by ASCT studies is the role of this procedure in patients with primary refractory DLBCL (i.e., those who are chemotherapy-induction failures). This group, in general, has a very poor prognosis. Several studies have attempted to assess the role of HDC, none of them in phase III. An initial report from the University of Nebraska indicated no patients with primary refractory disease were disease free beyond 1 year after HDC.192,193 These patients were not sensitive, however, to second-line chemotherapy. Other trials suggested better results could be obtained in patients responsive to second-line salvage programs.194–197 More recently, however, a large international phase III trial looking at salvage regimens demonstrated a 10% 3-year event-free survival in patients who had relapsed <12 months after induction therapy.198 The expert committee recommended against the use of HDC ASCT for newly diagnosed aggressive lymphoma patients with a partial response to induction chemotherapy without specifically addressing the issue of those patients who are nonresponders.
HDC and ASCT have generally not been successful in improving survival and curing patients with indolent disease (e.g., FL). These data have also been comprehensively reviewed recently.199 In brief, OS for indolent lymphomas does not appear to be improved with HDC/ASCT for relapsed disease. Late consequences, particularly the development of myelodysplasia or acute leukemia, are a real concern. The data on the use of allogeneic SCT are all from phase II trials, and, while promising, this procedure is still inhibited by the substantial treatment-related mortality, about 20% at 3 years for RIC transplants and 40% for myeloablative transplants. Thus, allogeneic SCT remains investigational.
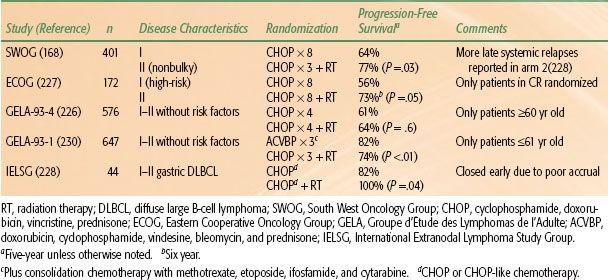
The role of radiotherapy in patients undergoing HDC with SCT, either autologous or allogeneic, is undefined. The authors have recently reviewed this issue.200 The rationale for RT lies in the observation that most treatment failures after HDC SCT occur at sites of initial involvement. As mentioned above, consolidation RT was employed in the landmark Parma trial. It is also commonly used at a number of institutions, usually directed at bulk disease sites present before the start of salvage chemotherapy, but with considerable interinstitutional variation and without a clear definition of what constitutes bulk disease. There are a number of phase II trials but no phase III trials addressing this issue. The majority of phase II trials do suggest benefit. The authors recommend doses of 20 to 30 Gy for those patients who have not received prior RT, depending on clinical circumstances and also dependent on whether or not TBI is planned as part of the conditioning regimen. Generally, it is preferable to irradiate prior to reinfusion of stem cells.
Immunotherapy
Perhaps the most promising new approach to the treatment of NHL has been the recent development of effective immunotherapy. The malignant lymphomas express a variety of surface antigens, most notably the B-cell antigen CD20. The ubiquitous presence of the CD20 antigen in many varieties of B-cell lymphomas led to the genetic engineering of a human chimeric anti-CD20 antibody rituximab. In contrast to prior murine derived monoclonal antibodies, rituximab is quite well tolerated in humans. Rituximab was the first antibody of any type to receive U.S. Food and Drug Administration (FDA) approval (1997) for the treatment of any human malignancy.
Numerous trials of rituximab have been carried out in virtually all B-cell lymphomas.201–207 Responses as a single agent are seen frequently in FL, CLL, MCL, and MZL. Although responses are infrequent in DLBCL, the addition of rituximab to the standard CHOP program significantly improves outcomes.183,184 Indeed, the addition of rituximab to chemotherapy for DLBCL represents the major advance of the past several decades in the systemic treatment of DLBCL. The effect is so substantial as to force a re-evaluation of prognostic factors as well as other adjuvant therapies (such as stem cell transplantation or radiotherapy) in the rituximab era.
Rituximab is also employed frequently in combination with chemotherapy for FL, both in the induction phase as well as for maintenance, and significantly improves both response rate and duration of response. Its effect on survival for FL is less clear. It has been combined with chemotherapy for MCL as well.204,208,209,210–211
In parallel with the development of rituximab, efforts were undertaken to link radioactive isotopes to anti-CD20 antibodies, in view of the known radiosensitivity of lymphomas. Currently two such radiolabeled anti-CD20 antibodies have been successfully developed: iodine-131 [131I] tositumomab (Bexxar) and yttrium-90 [90Y] ibritumomab tiuxetan (Zevalin). Both of these agents were FDA approved in 2002 and 2003, respectively. Both demonstrate significant antilymphoma activity, either alone or in combination with other chemotherapeutic regimens.212,213 They demonstrate efficacy in patients resistant to both chemotherapy and rituximab.214
Most of the experience has been gained with FL. The overall response rates are in the range of 80% with approximately one-third of patients achieving CR. In relapsed large cell lymphoma patients, response rates are somewhat lower (approximately 40%). In one trial of untreated FL patients very high response rates of 95% overall with 75% CR were achieved.215 In a phase III Canadian trial for advanced FL, consolidation 90Y-ibritumomab tiuxetan significantly improved CR rates (53% vs. 87%) as well as the median PFS (13.3 vs. 36.5 months).216 There is additional evidence to suggest efficacy of radioimmunotherapy as consolidation for aggressive histologies.217 The optimal timing of radioimmunotherapy, selection of appropriate patients, and integration into other available therapeutic modalities has not been established. These data have recently been reviewed by the Seattle group.218
TABLE 78.7 RANDOMIZED TRIALS EVALUATING CONSOLIDATION RADIATION THERAPY IN EARLY-STAGE DIFFUSE LARGE B-CELL LYMPHOMA