Localized Disease
Approximately 5% to 15% of patients with follicular lymphoma will have localized disease at diagnosis. These patients may experience prolonged disease-free survival with a watchand-wait approach, especially if they are free of disease after diagnostic biopsy.
49,50,51 The 10-year survival in a selected group of patients with stage I to II follicular lymphoma managed with a watch-and-wait approach at Stanford was estimated at 85%.
51 The median survival was 19 years, and 56% were still untreated after 10 years. This approach may be reasonable for some asymptomatic patients who do not desire therapy, are elderly, or have other medical conditions.
Involved-field radiation therapy for localized low-grade lymphoma is associated with 10-year relapse-free survival rates of approximately 50%.
52,53,54 It is unclear whether survival is improved with the addition of chemotherapy. Investigators from M.D. Anderson Cancer Center performed a prospective trial of combined-modality treatment for patients with stage I to II low-grade follicular lymphoma.
55 Patients received cyclophosphamide, vincristine, prednisone, and bleomycin (COP-bleo) or CHOP-bleo (COP-bleo and doxorubicin) combined with involved-field radiation. At 10 years, the time to treatment failure was estimated to be 72%, and overall survival was estimated at 80%. A prospective randomized British National Lymphoma Investigation (BNLI) trial found no improvement in disease-free survival or overall survival when chlorambucil was added to involved-field radiation therapy in patients with localized low-grade lymphoma.
56
Advanced Stage
Various treatment options are available for patients with follicular lymphoma who have advanced-stage disease at diagnosis. The National LymphoCare Study documented wide variation in management of US patients with newly diagnosed follicular lymphoma.
57 Nevertheless, the overall survival of patients with follicular lymphoma has improved and more than 70% of patients survive 10 years in the United States.
58,59Definite indications for therapy in patients with follicular lymphoma include rapidly progressing or symptomatic adenopathy or splenomegaly, symptomatic effusions, and cytopenias. In addition, patients frequently request therapy. A watch-and-wait approach is appropriate for asymptomatic patients, especially if they are elderly or have comorbid conditions.
60,61 There is little evidence that survival is shortened by deferral of treatment until patients are symptomatic. A trial from the National Cancer Institute randomized patients with advanced low-grade lymphoma to observation or to treatment with ProMACE-MOPP (prednisone, methotrexate, doxorubicin, cyclophosphamide, etoposide, mechlorethamine, vincristine, procarbazine, prednisone) combined with total nodal irradiation.
62 The 4-year disease-free survival was estimated at 51% following aggressive therapy as compared with 12% after observation (
P < .001), although overall survival advantages were not seen. A three-armed trial conducted by the French Groupe d’Etude des lymphomes de l’Adulte (GELA) cooperative group randomized follicular lymphoma patients with low tumor burden to treatment with observation, prednimustine, or interferon-
α.63 Significant differences in overall survival were not observed. Patients in another BNLI trial were randomized between oral chlorambucil and a watch-and-wait approach.
64 The median survival rates in the two groups were 6.7 and 5.9 years, respectively (
P = .44).
Single-agent cyclophosphamide or chlorambucil, or combination therapy with cyclophosphamide, vincristine, and prednisone (CVP), may be appropriate for some patients.
65 Numerous prospective-randomized trials have compared treatment with different chemotherapeutic regimens for patients with lowgrade lymphomas.
66,67,68,69,70,71 Although progression-free survival is
often increased with more aggressive therapies, significant overall survival advantages have not been demonstrated.
The purine analogs, fludarabine, 2-deoxycoformycin, and 2-chlorodeoxyadenosine, have single-agent response rates of 25% to 50% in previously treated patients with low-grade lymphomas.
72 The overall response rate was 65% when single-agent fludarabine was used for untreated follicular lymphoma.
73 The combination of fludarabine, mitoxantrone, and dexamethasone has an overall response rate of 70% to 90% for newly diagnosed follicular lymphoma patients.
74,75 Similar results have been seen with the combination of fludarabine and cyclophosphamide.
76,77A GELA trial randomized elderly patients between fludarabine, or to treatment with cyclophosphamide, doxorubicin, teniposide, and prednisone (CHVP) combined with interferon.
78 The 2-year actuarial survival was 62% in the fludarabine arm, as compared with 77% in the other arm (
P < .05). A phase III trial conducted by the European Organization for Research and Treatment of Cancer compared single-agent fludarabine with CVP for patients with stage III and IV low-grade non-Hodgkin lymphoma.
79 The response rate was higher in the fludarabine arm (70% vs. 52%;
P < .001), although there were no significant differences in failure-free survival or overall survival.
A prospective trial from M.D. Anderson Cancer Center compared the combination fludarabine, mitoxantrone, and dexamethasone with a 13-drug alternating triple therapy (ATT) regimen.
80 The 5-year failure-free survival was estimated at 41% with the fludarabine-based regimen, as compared with 50% for the ATT regimen (
P = .02). No significant difference in 5-year survival was noted, however. In another trial, the combination of fludarabine and mitoxantrone was compared with cyclophosphamide, vincristine, doxorubicin, and prednisone (CHOP).
81 The complete response rates were 68% and 42%, respectively (
P = .003), although overall survival was not significantly different. Another phase III trial compared fludarabine and mitoxantrone with CHVP in elderly patients with indolent non-Hodgkin lymphoma.
82 The 4-year failure-free survival was 42% in the fludarabine-treatment arm, as compared with 10% for patients treated with CHVP (
P = .0001). No significant differences in overall survival were noted.
Monoclonal Antibodies
Rituximab is a chimeric human-mouse anti-CD20 antibody that has revolutionized the treatment of follicular lymphoma. Phase II trials demonstrated response rates of approximately 50% in previously treated patients with follicular lymphoma.
89 Median response duration is approximately 12 months and prolonged remissions are seen, although no plateau in diseasefree survival is evident. Rituximab responses can be seen in chemotherapy-resistant patients and in patients who have relapsed after autologous hematopoietic stem cell transplantation (AHSCT). Patients who relapse after rituximab treatment will often respond to a second course of treatment.
90 Prolonged event-free survival has been observed when singleagent rituximab has been used as initial therapy for untreated patients.
91,92,93Randomized trials have demonstrated significant increases in response rate, progression-free survival, and overall survival when rituximab is combined with conventional chemotherapy regimens.
94,95,96,97 A German trial randomized patients with follicular lymphoma between treatment with CHOP and R (rituximab)-CHOP.
94 The estimated 2-year survival was 90% and 95%, respectively (
P = .016). Follicular lymphoma patients in an East German trial were randomized to treatment with mitoxantrone, chlorambucil, and prednisolone (MCP) or R-MCP.
95 The actuarial 4-year survival rates were 74% and 87%, respectively (
P = .0096). A third prospective trial for follicular lymphoma patients compared CVP with R-CVP.
96 The estimated 4-year survival rates were 77% and 83%, respectively (
P = .029). Although there is disagreement about the optimal upfront chemotherapy regimen for follicular lymphoma,
57 there is consensus that rituximab should be added to chemotherapy.
Additional rituximab following CHOP or fludarabine-based therapy can increase the complete response rate.
81 The use of maintenance rituximab for patients with follicular lymphoma improves outcomes with various regimens. A randomized Swiss trial with single-agent rituximab used in the upfront and relapse settings for patients with follicular lymphoma investigated the value of rituximab maintenance
93 administered at months 3, 5, 7, and 9. The median event-free survival was prolonged from 13 to 24 months with maintenance therapy (
P < .001). An intergroup trial randomized patients with follicular lymphoma to observation or maintenance rituximab following treatment with CVP.
98 Maintenance rituximab was administered weekly for 4 weeks, every 6 months (four courses). The estimated 3-year progression-free survival was 33% following CVP and 64% following CVP with maintenance rituximab (
P = 9.2 × 10
-8). Overall survival rates were estimated at 86% and 91%, respectively (
P = .08).
The question of whether maintenance rituximab is beneficial for patients with follicular lymphoma who receive rituximab with upfront chemotherapy was addressed in the PRIMA study.
99 Patients in this trial received R-CVP, R-CHOP, or fludarabine, cyclophosphamide, and mitoxantrone (R-FCM). Responders were then randomized between observation and maintenance rituximab administered every 8 weeks for 2 years. The estimated 2-year progression-free survival was 66% for the observation arm and 82% with maintenance rituximab (
P < .0001). Differences in overall survival were not observed. Rituximab has been approved for maintenance
therapy following upfront chemoimmunotherapy for follicular lymphoma. Rituximab increases the response rate with salvage chemotherapy for follicular lymphoma and prolongs progression-free survival.
100,101 Rituximab maintenance prolongs progression-free survival following salvage chemotherapy for follicular lymphoma.
101 A meta-analysis showed a significant improvement in overall survival when maintenance rituximab was used for patients with relapsed follicular lymphoma (hazard ratio 0.68).
102Additional anti-CD20 antibodies as well as antibodies directed at other antigenic targets are under development.
103 Ofatumumab is a monoclonal antibody that targets a novel CD20 epitope.
104 This agent is approved for the treatment of patients with refractory chronic lymphocytic leukemia. A phase I/II study demonstrated clinical activity with ofatumumab for patients with relapsed and refractory follicular lymphoma, even if they had previously received rituximab.
105The radiolabeled antibodies
90Y-ibritumomab tiuxetan and
131I-tositumomab are approved for patients with relapsed and refractory low-grade, follicular, or transformed B-cell lymphomas. Response rates of 50% to 80% have been observed for patients with follicular and other indolent lymphomas, including patients who were refractory to chemotherapy and rituximab.
106,107,108 Response rates may be higher than those of rituximab alone.
108 Radiolabeled antibodies have been used as initial therapy for follicular lymphoma.
109 Radiolabeled antibodies have also been tested in phase II trials following primary chemotherapy with CVP and CHOP.
110,111 In a phase III trial, follicular lymphoma patients who responded to primary chemotherapy were randomized between observation or to treatment with
90Y-ibritumomab tiuxetan consolidation.
112 Additional therapy with the radiolabeled antibody prolonged the median progression-free survival from 13.3 to 36.5 months (
P < .0001), although no overall survival differences were reported. This agent is now approved for use in untreated patients with follicular lymphoma who respond to upfront therapy.
New Drugs
Bendamustine administered as a single agent or in combination with rituximab yields response rates of 80% to 90% in patients with relapsed and refractory follicular lymphoma and other indolent subtypes.
113,114,115 In a prospective trial patients with untreated follicular lymphoma and mantle cell lymphoma were randomized between treatment with R-CHOP and R-bendamustine.
116 The median progression-free survival was 46.7 months following R-CHOP and was not yet reached for patients receiving bendamustine (
P = .0002). Patients in the bendamustine arm also had significantly less toxicity. The proteasome inhibitor bortezomib, administered alone or in combination with rituximab, is also active for patients with relapsed and refractory indolent lymphomas.
117,118 The immunomodulatory agent, lenalidomide, may also induce durable responses for patients with relapsed and refractory follicular lymphoma.
119 Ongoing trials are testing combinations of these newer agents.
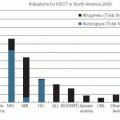
Hematopoietic Stem Cell Transplantation
Prospective randomized trials have investigated the role of AHSCT as part of initial therapy for patients with follicular lymphoma.
120,121,122,123 Although progression-free survival is prolonged, overall survival advantages have not been demonstrated. Only one of these trials utilized rituximab treatment,
122 and this approach cannot be recommended outside of a clinical trial. A single randomized trial performed prior to the introduction of rituximab showed that AHSCT was superior to conventional salvage chemotherapy for patients with chemotherapysensitive relapsed follicular lymphoma.
124 Additional reports have demonstrated that long-term progression-free survival can be observed in approximately 40% to 50% of patients with relapsed follicular lymphoma following AHSCT.
125,126,127,128 Allogeneic hematopoietic stem cell transplantation has the potential to cure patients with follicular lymphoma, although the morbidity and mortality may be high.
129