Nitric Oxide: The Vascular Biology of Nitric Oxide and Nitric Oxide Synthases
Bradley A. Maron
Thomas Michel
Nitric oxide (NO) is a ubiquitous, biologically versatile, and structurally unique molecule that is central to a diverse range of cell signaling processes that influence vascular function. The chemical properties of NO are ideally suited for participation in the inter- and intracellular reactions that modulate blood vessel relaxation.1,2 NO is distinctive among other endothelium-derived relaxing factors in several ways. NO is a small (30 Da) lipophilic gaseous molecule. These properties allow NO to easily traverse vascular endothelial and smooth muscle cell membranes and participate in intercellular signaling.3,4,5 Moreover, NO is a labile free radical that may react with a diverse range of molecules including thiols, heavy metals, and other biologically active free radicals such as superoxide anion. The relatively short half-life (5 to 10 seconds)6 of NO ensures a rapid and localized signaling effect,4,7 permitting NO to exert a tightly regulated, juxtacrine effect on the function of neighboring cells while also modulating intracellular signaling pathways. Importantly, the endothelial isoform of nitric oxide synthase (eNOS), which synthesizes NO, plays an important role in maintaining vascular homeostasis by virtue of the heterogeneous array of extracellular signals that modulate NO levels through influencing the eNOS gene, transcript, and protein. Posttranslational modifications to eNOS regulate its enzyme activity, subcellular targeting, and, importantly, the role of eNOS in vascular homeostasis; alterations in eNOS are implicated in the pathogenesis of several vascular disease states.
NITRIC OXIDE BIOCHEMISTRY
Targets of NO
NO produced by eNOS in vascular endothelial cells rapidly diffuses into adjacent vascular smooth muscle cells (VSMCs) and into the blood vessel lumen. The chemistry of NO allows for numerous reactions, including those with (a) proteins that contain metallic centers and/or sulfhydryl groups; (b) low molecular weight thiol compounds; (c) hemoproteins; or (d) other radicals.
Soluble Guanylyl Cyclase
In VSMCs and circulating platelets, the heterodimeric (α1/β1 or α1/β2) enzyme soluble guanylyl cyclase (sGC) is a critical NO target. NO-sGC signaling is essential for normal vascular reactivity and for suppression of abnormal platelet aggregation. NO binding to the heme prosthetic group of sGC disrupts a nitrogen-iron interaction, thereby freeing an axial histidine residue on the catalytically active β1 subunit of sGC. Nanomolar concentrations of NO may induce a 100-fold increase in the sGC-dependent conversion of cytosolic GTP to cGMP, which in VSMCs activates cyclic GMP-dependent protein kinase (also known as protein kinase G [PKG]) to promote vascular relaxation (discussed below). Diminished eNOS abundance and decreased enzyme activity can lead to reduced NO synthesis, leading to the attenuated activation of sGC and causing increased vascular tone and blood vessel injury. These alterations in eNOS bioactivity are implicated in the pathophysiology of important vascular diseases including stroke and myocardial infarction.8,9 Moreover, the potent vasodilatory effects of NO may be limited significantly owing to pathologic posttranslational modifications of sGC or other downstream signaling intermediaries. For example, oxidation of the heme moiety or key cysteinyl thiol(s) located near the NO-sensing pocket of sGC converts sGC to an NO-insensitive state and attenuates NO-dependent vasodilation.8,10 This model illustrates a key distinction in NO biology: NO bioavailability, which suggests the presence of NO levels that are sufficient to induce a biologic response, is not synonymous with NO bioactivity. The latter implies an appropriate interaction between NO and NO targets that results in a downstream functional effect on cellular physiology.
Formation of NO Adducts: Nitrosothiols
The relatively short half-life of NO has generated speculation regarding potential storage forms of NO that may serve to sustain adequate basal levels of NO and permit actions of NO to occur at targets distant from its sites of synthesis.11 The redox reactivity of nitrogen oxides with sulfhydryl groups is well characterized, and various nitrosothiols, including proteins covalently modified by a NO adduct, may represent a molecular “depot” for storage of NO in vivo.12 Both protein and nonprotein nitrosothiols have been found in cells and biologic fluids in vivo. Compared with a free plasma NO concentration of approximately 3 nM, the concentration of plasma nitrosothiol is approximately 1 µM, of which 85% is nitrosoalbumin.13,14,15 S-nitrosothiols are endogenously formed in both the cardiovascular and pulmonary systems,13,14 and micromolar concentrations of S-nitrosoglutathione have been isolated from the lung.
Nitrosothiols share a qualitatively similar pharmacodynamic profile with NO, such as stimulating sGC,11 inducing vascular relaxation and hypotension, and exerting antithrombogenic effects via inhibition of platelet aggregation.16 The molecular mechanism(s) by which endogenous nitrosothiols induce biologic effects remains speculative: nitrosothiols may undergo spontaneous nonenzymatic decomposition to release NO, induce transnitrosylation of other proteins, or directly exert nitrergic effects.17
Hemoglobin is proposed to be a nitrosoprotein with potential biologic relevance. While it is clearly established that hemoglobin has the ability to bind NO at its heme moieties, hemoglobin
may also undergo nitrosylation at Cys93 in the β globin protein to form a protein nitrosothiol. Although there is evidence to suggest that nitrosohemoglobin may release NO, the utility of exploiting NO binding by hemoglobin as a pharmacotherapeutic system for NO delivery to distal points in the vascular system remains under investigation.18 However, other mechanisms that utilize S-nitrosothiols for NO delivery have been tested in vivo. Through S-nitrosothiol linking, numerous NO molecules may be covalently bound to polyethylene glycol-conjugated bovine serum albumin, a macromolecule that is resistant to intermolecular disulfide bond formation. It has been demonstrated that intravenous injection of this compound in mice is associated with an increase in whole blood levels of the stable NO metabolite, nitrite, and a reduction in mean arterial blood pressure.19 The translation of this technology into an effective therapy remains the focus of ongoing investigations.20,21
may also undergo nitrosylation at Cys93 in the β globin protein to form a protein nitrosothiol. Although there is evidence to suggest that nitrosohemoglobin may release NO, the utility of exploiting NO binding by hemoglobin as a pharmacotherapeutic system for NO delivery to distal points in the vascular system remains under investigation.18 However, other mechanisms that utilize S-nitrosothiols for NO delivery have been tested in vivo. Through S-nitrosothiol linking, numerous NO molecules may be covalently bound to polyethylene glycol-conjugated bovine serum albumin, a macromolecule that is resistant to intermolecular disulfide bond formation. It has been demonstrated that intravenous injection of this compound in mice is associated with an increase in whole blood levels of the stable NO metabolite, nitrite, and a reduction in mean arterial blood pressure.19 The translation of this technology into an effective therapy remains the focus of ongoing investigations.20,21
In addition to functioning as a storage reservoir of NO, NO-protein adducts may serve as an important posttranslational modification of biologically active proteins.22 For example, a novel molecular mechanism by which S-nitrosylation influences vascular tone has recently been reported. Zhou et al. demonstrated that activation of angiotensinogen, a key vasoactive octapeptide that regulates blood pressure in mammals, is dependent, in part, on oxidation-induced disulfide bond formation between Cys18 and Cys138. Under reduced conditions in which this disulfide bond is broken, however, S-nitrosylation at these critical cysteines may occur. In turn, this induces a stereometric change to angiotensinogen that prevents Cys18-Cys138 disulfide bond formation, and thereby attenuates the angiotensinogen-mediated hypertensive response in vivo.23
Nitrosylation pathways may also regulate proteins associated with hemostasis. For example, S-nitrosoglutathione may interact with fibrinogen and impair fibrin cross-linking, without chemically reacting with fibrinogen but possibly by inducing a conformational change in fibrinogen.24 Nitrosylation of factor XIII may be an antihemostatic mechanism that could attenuate fibrin cross-linking and render a nascent clot more susceptible to fibrinolysis.25 While nitrosylation of tissue-type plasminogen activator (S-NO-t-PA) has no effect on its interaction with fibrin or the catalytic activation of plasmin, S-NO-t-PA may act as a nitrosothiol carrier of NO to inhibit platelets and cause vasodilatation at sites of thrombus where the local endothelium may be dysfunctional and producing insufficient quantities of NO.26
NO Adducts and Reactive Oxygen Species
Formation of NO adducts may be affected by the cellular redox potential, and in turn may influence the nature of redox-active species (e.g., reactive oxygen species) present in the vascular wall and influence NO bioavailability. The most prominent example of this phenomenon is the formation of peroxynitrite through the reaction of NO and superoxide. Although this process may be a mechanism for detoxifying superoxide, it also leads to decreased levels of bioavailable NO through several mechanisms. First, generation of peroxynitrite results in depletion of the NO substrate. Second, peroxynitrite may react with nitrite to form peroxynitrate, thereby decreasing levels of nitrite, which is a key substrate for NOS-independent NO synthesis.10 Finally, peroxynitrite itself is a chemically reactive species that may adversely modify cellular proteins involved in NO-dependent cell signaling pathways.
SOURCES OF NITRIC OXIDE
Nitric Oxide Synthases
NO is synthesized endogenously in mammalian tissues by a family of three nitric oxide synthase (NOS) isoforms, each the product of a distinct gene. The mammalian NOSs are homodimeric enzymes containing an N-terminal heme-binding domain and a C-terminal reductase domain, separated by a calmodulin-binding domain. The C-terminal domains of the three proteins in the mammalian NOS enzyme family are structurally similar to cytochrome P450 reductases.27,28,29 The N-terminal domain of NOS, also known as the oxygenase domain, contains binding sites for heme (protoporphyrin IX), tetrahydrobiopterin, and the substrate L-arginine.28 The C-terminal flavoprotein oxidoreductase domain binds flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), and NADPH.30 As these two functions are often the products of separate polypeptides, the combination of the oxygenase and reductase functions of NOS suggests that the prototypical NOS protein evolved by gene fusion, with a binding site for the Ca2+-binding protein calmodulin separating the N- and C-terminal domains.
eNOS Catalysis
Under basal conditions, electron transfer between NOS domains is suppressed; however, it appears that calmodulin promotes the flow of electrons from the reductase to the oxygenase domains.31 The fact that NOS binds five redox-active cofactors—more than any other enzyme known—suggests an intricate catalytic mechanism. NOS catalyzes the five-electron oxidation of the guanidine nitrogen of L-arginine, although reactions of NADPH and flavoproteins typically involve an even number of electrons. NOS catalysis proceeds in two distinct monooxygenation steps, each involving electron transfer to molecular oxygen: in the first step, one mole of NADPH transfers, via FAD and FMN,32 two moles of electrons to a heme-bound oxygen, which reacts with L-arginine to form one mole each of omega-N-hydroxy-L-arginine and water.33 In the second step, one half mole of NADPH transfers electrons to a second heme-bound oxygen, which reacts with omega-N-hydroxy-l-arginine to release one mole each of NO and L-citrulline.
NOS Isoforms
Of the three distinct NOS isoforms that have been identified, two are constitutively expressed in their archetypal cells of origin: eNOS in vascular endothelial cells and neuronal NOS (nNOS) in neurons. As the name implies, inducible NOS (iNOS) is an inducible isoform found in a broad range of immunoactivated cells (see Table 40.1). Such terminology is somewhat misleading, however, because NOSs are expressed in a diverse range of tissues other than endothelium, neurons, and immunoactivated cells. Furthermore, the designation of constitutive and iNOS expression patterns are not strictly maintained, as nNOS and eNOS genes can undergo tissue-specific transcriptional regulation. Moreover, while eNOS is constitutively expressed, it may be induced by extracellular mediators. The overall catalytic scheme of the three mammalian NOS isoforms is conserved, and the members of this gene family share similar gene structure and exhibit 50% to 60% mammalian amino acid sequence homology,34,35,36 as well as nearly identical locations of cofactor binding sites. There is considerable heterogeneity among the isoforms at the N-termini. For example, nNOS contains an additional sequence of approximately 290 amino acids not found in either eNOS or iNOS. Moreover, the N-terminus of eNOS is distinguished from nNOS and iNOS by
the presence of consensus sites for acylation, which are thought to determine eNOS subcellular localization.37
the presence of consensus sites for acylation, which are thought to determine eNOS subcellular localization.37
Table 40.1 Cellular and tissue localization of NOS expression | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
All three NOS isoforms catalyze the same chemical reaction and have similar redox cofactor requirements. A major difference lies in the isoforms’ differential affinity for calmodulin: although all three NOS isoforms are dependent on calmodulin binding for activity, there are important differences in the calcium dependence for calmodulin binding. Whereas eNOS and nNOS require elevated intracellular calcium levels in order to bind calmodulin and thereby become activated, the affinity of iNOS for calmodulin is sufficiently high that iNOS fully binds calmodulin at the ambient intracellular calcium concentrations present in resting cells, and typically iNOS is persistently activated once the protein is synthesized.38
VASCULAR FUNCTIONS OF NOS-DERIVED NITRIC OXIDE
NOS, NO, and the Blood Vessel Wall
NO is an essential endogenous vasodilator, platelet inhibitor, and natural antithrombotic agent. Numerous experiments in both animal models and humans indicate that the physiologic role of eNOS is to produce NO in response to diverse extracellular stimuli and thereby maintain both a baseline level of systemic vasodilatation and establish vasomotor tone. In addition to its anticoagulant activity, endothelium-derived NO also attenuates endothelial proliferation and leukocyte adhesion. In turn, endothelial dysfunction that occurs as a consequence of decreased eNOS-derived NO may be a central mechanism involved in the pathobiology of vascular diseases characterized by vasoconstriction, hypertension, and a prothrombotic state.
The functional consequences of abnormal eNOS activity have been extensively studied. Both knockout and overexpression experiments suggest a role of eNOS and NO in maintaining basal vascular homeostasis. eNOSnull mice are hypertensive, demonstrate increased leukocyte-endothelial adherence, and diminished bleeding time.39,40,41 These eNOSnull mice also develop pulmonary hypertension, hypoxia, and right ventricular hypertrophy.42 Conversely, transgenic mice that overexpress eNOS show increased basal levels of NO and cGMP, associated with decreased blood pressure.43
Compensatory Bioactivity of NOS Isoforms
Animals genetically engineered to express various combinations of NOS isoforms (including the absence of all three isoforms), provide insight into the compensatory bioactivity of these enzymes in vivo. For example, pyramidal tissue-initiated long-term potentiation (i.e., sustained signal transmission) of hippocampal neurons is abolished in doubly mutant n/eNOSnull mice, but preserved in mice singly mutant for nNOS or eNOS.44 Although eNOS protein expression levels are elevated in pyramidal tissue relative to central nervous tissue overall, the ratio of eNOS: nNOS protein expression in pyramidal tissue is quite low.45 Thus, the observation that eNOS-derived NO levels are sufficient to sustain long-term potentiation suggests that functional efficiency of NOS varies between enzyme isoform and/or with respect to tissue type.
Studies performed in mice lacking all three isoforms (e/i/n NOSnull “triple knockout” mice) provide insight into the importance of NOS-independent mechanisms of NO synthesis in vivo. This “triple knockout” model also provides an informative model for understanding compensatory actions in the absence of NOS isoforms. Although the e/i/n NOSnull mice develop normally, they demonstrate significantly decreased rates of survival compared to wild type counterparts.46 Interestingly, basal levels of NO are detectable in vascular tissue harvested from the triple-null animals, which probably reflects the conversion of dietary or bacteriaderived nitrite to NO.47 In fact, there is accumulating evidence to suggest that nitrite (NO2–) and nitrate (NO3–), present in plasma of mammals via metabolism of endogenous NO, or, in the case of NO3–, from dietary intake of foods rich in inorganic nitrate, are biologically important through their recycling to NO.48 A typical Western diet, for example, is rich in NO3–, which is readily absorbed in the gastrointestinal tract. Although mainly excreted in urine, the balance (as much as 40%) of circulating NO3– is ultimately excreted into the saliva.49 There, NO3– may be converted to NO2– by endogenous oral flora in a reaction that requires nitrate reductase. In the circulation, NO2– may be reduced by deoxyhemoglobin and/or deoxymyoglobin to form bioavailable NO.50 Cycling of NO, NO2–, and NO3– may be facilitated in the presence of xanthine oxidoreductase, hypoxia, or, even by NOS.48
In addition to the implications of NOS-independent NO synthesis on vascular function that are gathered from NOS knockout rodent models, it has been demonstrated previously that
dietary NO3– loading or the administration of inorganic NO2– via nebulizer or intravenous access influences vascular tone to decrease blood pressure. This has been demonstrated in the peripheral, pulmonary, and cerebrovascular circulations.51,52,53,54,48
dietary NO3– loading or the administration of inorganic NO2– via nebulizer or intravenous access influences vascular tone to decrease blood pressure. This has been demonstrated in the peripheral, pulmonary, and cerebrovascular circulations.51,52,53,54,48
As expected, the triple null mice exhibit many of the features associated with the absence of single NOS isoforms. For example, they have equivalent rates of hypertension and bradycardia as compared to eNOS-deficient mice. However, the loss of more than one isoform results in pathophysiologic changes not observed in singly NOSnull mice. For example, the number of disrupted NOS genes appears to positively correlate with the development of hypotonic polyuria, polydipsia, and renal responsiveness to vasopressin (i.e., nephrogenic diabetes insipidus).46 The development of nephrogenic diabetes insipidus in triply NOSnull mice may be linked with decreased NO-dependent cGMP formation and membrane insertion of aquaporin-2 in renal collecting ducts, two biomolecular processes that regulate urine tonicity.46
Despite the inhibitory effects of eNOS-derived NO on pathobiologic processes linked to the development of coronary artery disease, such as promoting vasodilation and inhibiting platelet aggregation and leukocyte adhesion, eNOS-deficient mice do not spontaneously develop arteriosclerosis. In contrast to eNOSnull mice, nNOS-deficient mice demonstrate elevated rates of metabolic syndrome, vascular lesion formation, and spontaneous myocardial infarction while iNOS deficiency is linked to atherogenesis.47 These findings suggest that nNOS and/or iNOS may play a vascular-protective, compensatory role under conditions in which eNOS levels are diminished.
eNOS Uncoupling
Synthesis of NO by eNOS is dependent upon the transfer of electrons from NADPH to FAD and then to FMN in a reaction that occurs in the reductase domain of the eNOS dimer complex. Tetrahydrobiopterin (BH4) is an eNOS cofactor required for electron donation from FMN to heme on the oxygenase monomer of eNOS that catalyzes the conversion of L-arginine + O2 → L-citrulline + NO.55 However, under conditions in which levels of BH4, L-arginine, or NADPH are depleted, the eNOS dimer “uncouples” and oxygen is converted to superoxide in preference to NO (FIGURE 40.1).
Decreased levels of bioavailable NO that occur as a consequence of eNOS uncoupling is a critical downstream effect of increased vascular oxidant stress, and is linked to the pathobiology of vascular dysfunction in hypertension, diabetes mellitus, hypercholesterolemia, and atherosclerosis.56 For example, eNOS uncoupling is postulated to be a mechanistic link between hyperaldosteronism and systemic hypertension. In peripheral blood vessels, elevated levels of aldosterone are associated with a vasculopathy characterized by activation of NADPH oxidase(s) that generates vascular endothelial superoxide formation. In turn, superoxide may oxidize BH4 to BH3 that results in eNOS uncoupling, decreased eNOS-dependent NO synthesis, and impaired endothelium-dependent vascular relaxation.57
In mice treated with streptozotocin to induce diabetes via pancreatic β-cell destruction, mouse aortas demonstrate increased total eNOS expression levels that are associated with diminished BH4 and low levels of eNOS dimer formation.58 Vascular dysfunction despite eNOS protein overexpression has been observed under a variety of experimental models of increased vascular oxidant stress. This paradox appears to reflect the effects of accelerated BH4 consumption by increased eNOS activity, which overall contributes to further superoxide formation by uncoupled eNOS, diminished levels of bioavailable NO, and worsening endothelium-dependent vascular relaxation.56,59
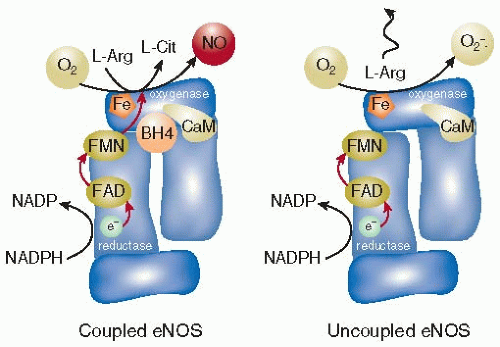 FIGURE 40.1 eNOS catalysis under normal conditions and in the uncoupled state. The synthesis of NO and L-citrulline (L-Cit) from molecular oxygen (O2) L-arginine (L-Arg) by eNOS requires five cofactors that permit the flow of electrons from the reductase domain to the oxygenase domain. NADPH donates an electron to the FAD-FMN complex. FAD/FMN facilitates the transfer of an electron to the iron of heme on the oxygenase domain, while calmodulin is believed to promote the flow of electrons from the reductase to the oxygenase domains. This, and the donation of an electron and proton from tetrahydrobiopterin (BH4) to O2/L-Arg are necessary for the synthesis of NO/L-Cit. In turn, if levels of the eNOS cofactors BH4 or NADPH, or the substrate L-Arg are insufficient, eNOS may become “uncoupled.” Under these conditions, O2 is converted to the reactive oxygen species superoxide (•O2–) in preference to NO. CaM, calmodulin; Fe, iron; e–, electron. (From Schulz E, Jansen T, Wenzel P, et al. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid Redox Signal 2008;10:1115-1126.) |
Other important mechanisms in addition to BH4 depletion are implicated in the uncoupling of eNOS. For example, the endogenous NOS inhibitor asymmetric dimethylarginine (ADMA) is linked to eNOS-derived superoxide formation in pulmonary artery endothelial cells,60 possibly by competitively inhibiting the binding of L-arginine. Circulating ADMA levels positively correlate with cardiovascular mortality.61 Interestingly, the ADMA-degrading enzyme dimethylarginine dimethylaminohydrolase (DDAH) is redox sensitive: under conditions of increased vascular oxidant stress, inhibition of ADMA by DDAH is decreased resulting in elevated plasma ADMA levels.61,62,63
Genetic Etiologies of eNOS Dysfunction
A number of human studies have reported increased prevalence of specific eNOS polymorphisms in hypertensive cohorts.64 For example, one major eNOS polymorphism is Glu298Asp, which has been associated with decreased NOS activity in the renal artery,65 reduced basal production of NO,66 coronary vasospasm,67 essential hypertension,68,69 hypertension in pregnancy,70,71 high-altitude pulmonary edema,72 as well as elevated risk of atherosclerosis73 and possible myocardial infarction.74,75
However, the mechanism by which this genetic polymorphism alters eNOS function remains to be determined.
However, the mechanism by which this genetic polymorphism alters eNOS function remains to be determined.
NO and Vascular Smooth Muscle Relaxation
NO produced in the endothelium stimulates sGC in VSMCs to produce cGMP. Increase in cGMP functions as a potent second messenger to promote vascular smooth muscle relaxation by activating cGMP-dependent PKG-myosin light chain phosphatase (MLCP) signaling, as well as by regulating intracellular calcium levels (FIGURE 40.2). The latter response may be mediated by the effects of cGMP on voltage-gated calcium channels, sodium/calcium exchangers, calcium-ATPase, phospholamban (PH), the IP3 receptor, and G proteins.76 Alternatively, cGMP may independently regulate monovalent cation channels to affect intracellular calcium levels.
NO may also influence vasomotor tone through cGMP-independent pathways, including the activation of calciumdependent potassium channels, which serve to hyperpolarize vascular smooth muscle.77 NO can also rapidly and reversibly inhibit cytochrome c oxidase (complex IV). Additional evidence suggests that prolonged exposure to NO may persistently inhibit other heme or iron-sulfur cluster enzymes of the respiratory chain including aconitase, NADH-ubiquinone oxidoreductase (complex I), and succinate-ubiquinone oxidoreductase (complex II).78 The rapid, reversible inhibition of cytochrome c oxidase is believed to provide a mechanism whereby NO may limit cell respiration and available cellular ATP, thereby attenuating smooth muscle contraction and causing passive vasodilatation.78,79 Longer-term inhibition of other enzymes in the respiratory chain may lead to the suppression of key metabolic pathways and to pathologic changes in VSMCs.78
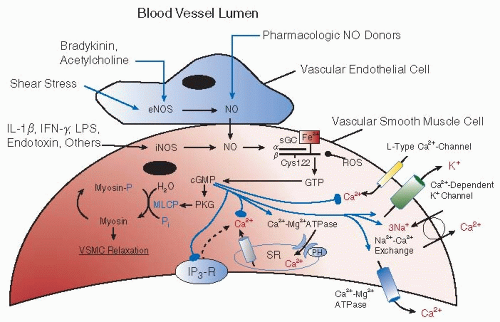 FIGURE 40.2 NO-induced cGMP formation modulates VSMC relaxation. In vascular endothelial cells, shear stress and/or agonist-mediated activation of a broad range of cell surface receptors increases eNOS activity and results in NO synthesis. NO derived from eNOS and/or pharmacologic donors may traverse the vascular endothelial cell membrane into the vascular smooth muscle cell (VSMC) layer. Alternately, induction of iNOS by various cytokines and/or endotoxin increases levels of bioavailable NO in VSMCs. The interaction of NO with soluble guanylyl cyclase (sGC) results in the conversion of cytosolic GTP to cGMP. Importantly, oxidative modification of sGC at the heme site or at key cysteinyl thiols (e.g., Cys122) on the catalytically active β1-subunit may impair NO-sensing by sGC and decrease conversion of GTP to cGMP. Protein kinase G (PKG)-dependent activation of myosin light chain phosphatase (MLCP) by cGMP induces VSMC relaxation. Additionally, cGMP modulates VSMC relaxation by decreasing intracellular levels of Ca2+. This occurs via cGMP-induced inhibition of Ca2+ mobilization through L-type Ca2+ channels or the inositol triphosphate-3 receptor (IP3-R) located on either the plasma membrane or in the sarcoplasmic reticulum (SR). In turn, cGMP also decreases intracellular Ca2+ levels by stimulating Ca2+-dependent K+ exchange, Na2+-Ca2+ exchange, Ca2+-Mg2+ ATPase(s), and phospholamban (PH) in the SR. Arrows and circular heads indicate agonist and inhibitory effects, respectively. eNOS, endothelial nitric oxide synthase; iNOS, inducible nitric oxide synthase; ROS, reactive oxygen species; LPS, lipopolysaccharide. (Adapted from Lucas KA, Pitari GM, Kazerounian S, et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev 2000;52(3):375-414. Ref. 238.) |
NO and Vascular Smooth Muscle Proliferation
NO inhibits the migration and proliferation of VSMCs.80 Pharmacologic NO donors exhibit antiproliferative effects in vitro,81 possibly by attenuating activity through activation of the cGMP-dependent protein tyrosine phosphatase, or “PTP-PEST.”82 Elevated levels of cGMP also activates cAMP-dependent protein kinases, which may account, in part, for the antiproliferative effect of NO (see below for additional details on NO and cAMP-dependent protein kinase).83
Antiproliferative characteristics of NO-donating drugs may also have an antiatherogenic effect in healthy endothelium vis-àvis preventing neointimal formation in vascular smooth muscle.
However, endogenous NO signaling pathways are also linked to migration84 and proliferation85 of VSMCs, as well as vascular endothelial growth factor (VEGF)-NO signaling-associated angiogenesis. Despite this and other similar observations, the preponderance of data currently available suggests that the attributable contribution of NO to the development of atherosclerosis is small, and is offset significantly by the proven beneficial effects of NO on maintaining normal endothelial function.
However, endogenous NO signaling pathways are also linked to migration84 and proliferation85 of VSMCs, as well as vascular endothelial growth factor (VEGF)-NO signaling-associated angiogenesis. Despite this and other similar observations, the preponderance of data currently available suggests that the attributable contribution of NO to the development of atherosclerosis is small, and is offset significantly by the proven beneficial effects of NO on maintaining normal endothelial function.
NO and Vascular Apoptosis
NO is a key mediator of apoptosis in various cell types including vascular endothelial and VSMCs.86 The effect of NO on apoptosis is concentration-dependent: low levels of NO are protective against apoptosis, whereas elevated levels of NO promote apoptosis and are cytotoxic.87 This provides a framework for interpreting the variable effects of each NOS isoform on the cell cycle. For example, iNOS induction in VSMCs or macrophages, which is associated with apoptosis, tends to generate significantly higher levels of NO compared with eNOS. In turn, eNOS exerts a predominantly antiapoptotic effect in vascular endothelial cells.
The mechanisms whereby NO inhibits apoptosis in the endothelium appear to be independent of cGMP and may involve the increased expression of heat shock proteins,88 inhibition of apoptotic kinase cascades, as well as inhibition of a family of proapoptotic cysteine proteases (termed caspases), probably via cysteine sulfhydryl nitrosylation and/or tyrosine nitrosation.89,90 Shear stress over the endothelial layer, which is an important determinant of basal NO synthesis by eNOS, may be an important antiapoptotic factor that serves to protect the endothelium against apoptosis in the initial stages of atherogenesis.91
The ability of iNOS-derived NO to exert proapoptotic and antiproliferative effects on vascular smooth muscle may contribute to the putative role of iNOS in vascular wall remodeling.92 NO-induced apoptosis of VSMCs may also be important in atherogenesis and plaque instability.93,94 The proapoptotic functions of iNOS-derived NO also appear to be cGMP-independent, and may relate to NO-induced oxidative damage of cytosine residues, which results in DNA fragmentation.95 Other mechanisms that have been proposed include: increased formation of peroxynitrite; activation of caspases; activation of poly(ADP ribose) polymerase; and increased mitochondrial permeability. Some evidence indicates an upregulation of p53 and/or p73 protein in apoptotic cells, and experiments demonstrate vascular smooth muscle from p53null mice are specifically more susceptible to NO-induced apoptosis as compared to wild type.96,97
Endothelial expression of iNOS may also induce apoptosis in endothelium in diabetic vasculopathy, where circulating glycosylated albumin derivatives have been reported to upregulate iNOS activity in small vessels.98 Additionally, expression of iNOS has been suggested as a mechanism of neuronal apoptosis that contributes to cardiovascular autonomic depression in septic shock.99
eNOS and Leukocyte-Endothelial Interactions
eNOS-derived NO exerts broad anti-inflammatory and antithrombogenic effects that arise in large part by the inhibition by NO of leukocyte-endothelial interactions. NO downregulates expression of P-selectin on endothelial cells.100 NO-mediated inhibition of P-selectin expression prevents the entire process of leukocyte recruitment. NO may also limit the expression of leukocyte adhesion molecules such as L-selectin or CD11/CD18.101 Since superoxide in the endothelium and leukocytes can promote oxidant-dependent adhesion, the inhibitory effects of NO on cell adhesion may be mediated in part by detoxification of superoxide.102 Inhibition of eNOS enzyme activity may result in the enhanced adherence of leukocytes to the vascular endothelium.102 Since monocyte adhesion may be one of the initial events in atherogenesis, eNOS may function as an antiatherogenic agent in part by inhibiting recruitment of leukocytes to endothelium. eNOS enzyme inhibition by arginine analogs also leads to increases in vascular permeability, extravasation of plasma proteins, and microvascular edema, involving both cGMP-dependent103 and cGMP-independent pathways of vascular permeability.104 These increased vascular permeability responses may be mediated in part by the increased adherence of leukocytes to endothelium in the absence of NO.105
eNOS and Blood Platelets
The same eNOS isoform originally described in vascular endothelium is also expressed in blood platelets, but the vascular endothelium is more significant quantitatively as a source of NO.106,107 Endothelial cell-derived NO is important to the regulation of platelet function, and is a key determinant of the balance between physiologically appropriate hemostasis and pathologic thrombosis. Nevertheless, platelet-derived NO from endogenous platelet eNOS may also play an important role in inhibiting platelet adhesion,39,80,108 aggregation,80 and activation. NO may affect platelets through several interrelated signaling pathways that variously involve cGMP, calcium, eicosanoids, and phosphoinositides as key effectors of the NO response.
Platelet adhesion to the endothelium depends on a number of adhesion molecules expressed on both cells, including intercellular adhesion molecule (ICAM)-1 and α/β 3 integrin on the endothelium, and β1 integrin, P-selectin, glycoprotein Ib, and glycoprotein IIb/IIIa on platelets.109 NO released by nitrovasodilator drugs inhibits expression of P-selectin on platelets.110 In addition to antagonizing platelet-endothelial interactions, the downregulation of P-selectin also inhibits platelet-leukocyte interactions. A PKG-modulated pathway also disrupts activation of GP IIb/IIIa, one of the more critical mechanisms of platelet adhesion to the endothelium. In this pathway, cGMPactivated PKG phosphorylates both phospholipase C and vasodilator-simulated phosphoprotein,111 which results in a decline in intraplatelet calcium. cGMP can also lower intraplatelet calcium by inhibiting release of intracellular calcium stores and shuttling more calcium out of the cell while limiting extracellular calcium reabsorption.108 Low calcium hinders protein kinase C signaling, hindering the activation of GP IIb/IIIa.110 The GP IIb/IIIa receptor is also necessary for aggregation of platelets in nascent thrombi by cross-linking platelets and fibrin. The potent platelet aggregating effects of thrombin are inhibited by NO112 from eNOS, which may also be activated by insulin.113 In platelets, insulin stimulates the Akt-mediated phosphorylation of eNOS Ser1177 via phosphoinositide-3-kinase (PI3K) and AMP-activated protein kinase (AMPK) pathways.113 In addition to insulin’s direct effects on eNOS in endothelial cells, insulin may also induce vasodilatation by activating platelet eNOS and
PKG,114 resulting in release of vesicles containing the vasoactive substances ATP, adenosine, and serotonin, which may in turn activate eNOS.115 The lipid mediator sphingosine-1-phosphate (S1P) is involved in a signaling between platelets and the endothelium: activated platelets secrete S1P, which stimulates activation of eNOS, thus releasing NO into the vascular lumen, which serves to attenuate platelet activity.
PKG,114 resulting in release of vesicles containing the vasoactive substances ATP, adenosine, and serotonin, which may in turn activate eNOS.115 The lipid mediator sphingosine-1-phosphate (S1P) is involved in a signaling between platelets and the endothelium: activated platelets secrete S1P, which stimulates activation of eNOS, thus releasing NO into the vascular lumen, which serves to attenuate platelet activity.
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


