Fig. 23.1
Clinical presentations of neuroblastoma. (a) Evidence of left-sided ptosis in a child with high-level thoracic tumor. This tumor frequently present with Horner’s syndrome (ptosis, miosis, and exophthalmos). (b) High thoracic neuroblastic tumors seen on computed tomography (CT scan). (c) Lower thoracic tumors surrounding the airway and likely intraspinal extension noted.
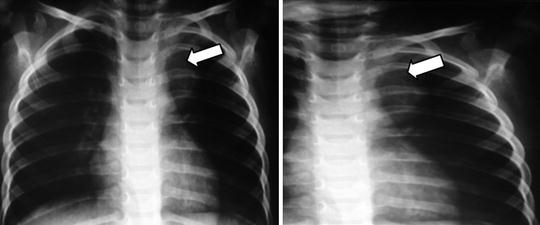
Fig. 23.2
Mediastinal tumors may results in respiratory distress, although those found in the lower mediastinum may be asymptomatic and discovered incidentally on routine radiographs obtained for unassociated medical reasons
The majority of children with neuroblastoma present with disseminated disease at diagnosis, with metastasis to bone marrow, bone, liver, lymph nodes, and less frequently lungs and central nervous system [25, 26]. Disseminated disease is usually associated with nonspecific symptoms, including fever, pallor, and anorexia and bone pain with subsequent changes in mood and refusal to ambulate. Disseminated neuroblastoma may present with several classic signs, such as infiltration of periorbital bone with proptosis and ecchymosis. A small group of infants with stage 4S may present with multiple bluish, painless, subcutaneous nodular lesions. Occasionally, these infants have massive hepatomegaly leading to coagulopathy, respiratory compromise, and renal insufficiency [27].
Tumor extension and growth within the foramen of the vertebrae with spinal compression are seen in approximately 5–15 % of young children with paraspinal tumors, resulting in symptomatic weakness, paralysis, bowel and/or bladder dysfunction, and radicular pain [28].
There have been two reported paraneoplastic syndromes associated with neuroblastoma. Opsoclonus-myoclonus is characterized by rapid multi-directional movements of the eyes (opsoclonus), muscle twitching/jerking (myoclonus), and thalamic ataxia and occurs in 1–2 % of the patients [29]. The exact mechanism of this autoimmune reaction and the reasons for its association with severe neurological sequelae is unknown. Another paraneoplastic syndrome is VIP syndrome; the tumors can secrete intestinal peptide vaso-active (VIP), which leads to chronic diarrhea, failure to thrive, hypokalemia, and dehydration. The VIP-secreting tumors are often ganglioneuroblastoma or ganglioneuroma and have an overall favorable prognosis [30].
Diagnostic Evaluation
A careful and thorough physical examination is critical; blood pressure for hypertension, complete neurologic examination to detect Horner’s syndrome, symptoms of spinal cord compression or abnormal movements such as opsoclonus, myoclonus, or ataxia to indicate OMS, skin nodules, or hepatomegaly for 4s disease, and proptosis to indicate a high-risk patient with orbital metastasis, thoracic, abdominal, or pelvic mass. Baseline laboratory studies should include CBC with differential and platelet, liver function tests, creatinine, serum LDH, ferritin, and urinalysis [29, 31]. The diagnosis of neuroblastoma requires the involvement of pathologists who are familiar with childhood tumors. Some neuroblastomas cannot be distinguished from other small round blue cell tumors of childhood, such as lymphomas, primitive neuroectodermal tumors, and rhabdomyosarcomas, using conventional light microscopy and routine H&E staining. Evidence for sympathetic neuronal differentiation may be demonstrated by immunohistochemistry, or by finding elevated levels of urine catecholamine metabolites, such as vanillylmandelic acid (VMA) or homovanillic acid (HVA), although 5–10 % of neuroblastomas do not secrete catecholamines [32].
Surgical Procedure
Surgical management of a child with suspected neuroblastoma should be thoroughly reviewed and discussed with the pediatric oncology team prior to taking the child to the operating room. In general, a limited initial open biopsy to obtain an adequate specimen for diagnostic and analysis of available prognostic markers should be considered [33]. Extensive upfront surgical resection and surgical laminectomy should be limited as this may result in delay in appropriate chemotherapy and potentially result in avoidable surgical complications [34].
Evaluation of the Bone Marrow
Disseminated disease is predictive and prognostic of poor outcome in children with neuroblastoma. Its accurate and sensitive assessment can facilitate optimal treatment decisions. The International Neuroblastoma Risk Group (INRG) Task Force has defined standardized methods for the determination of minimal disease by immunocytology and quantitative reverse transcriptase-polymerase chain reaction using disialoganglioside GD2 and tyrosine hydroxylase mRNA, respectively. One of the most common sites of metastasis in high-risk neuroblastoma is the bone marrow. Due to the heterogeneity of the bone marrow infiltration, it is critical to obtain multiple biopsies to detect neuroblastoma clusters. Unfortunately, the cytological and histological techniques lack sensitivity and standardization. Recently, the INRG group published its consensus criteria for the detection of neuroblastoma cells in bone marrow, peripheral blood (PB), and peripheral blood stem cells [35]. The objective of the International Risk Group Classification (INRG) is to develop standardized procedures to ensure the consistency of the results and allow comparisons between institutions.
Radiological Procedures
Treatment for children with neuroblastoma is guided by appropriate disease staging at time of initial diagnosis. Therefore, clinical staging should include:
1.
CT/MRI of the primary and metastatic sites.
2.
Bone scan.
3.
I123 metaiodobenzylguanidine (MIBG) (preferable over bone scan) or positron emission tomography (PET) scan.
CT Scan/MRI
CT scan and/or MRI of the chest, abdomen, and pelvis are considered the standard modalities for evaluation of solid tumor malignancies in children; additional areas of imaging may be guided based on presentation.
MRI is most effective in the detection of tumor extension into the epidural or leptomeninges and may provide a better definition of tumor extent for surgical planning. There has been an increased trend and preference for use of MRI, in an effort to limit the potential risks of secondary malignancies from radiation exposures of CT scans.
MetaIodobenzylguanidine and Positron Emission Tomography
The MIBG scan is a very sensitive and specific imaging modality for neuroendocrine tumors, particularly neuroblastomas. This scan aids in detection of primary and metastatic sites of neuroblastoma and response to therapy. Current data suggest that 123I-MIBG scan is preferred to the 131I-MIBG and Tc-99 bone scan, because of greater sensitivity, improved image quality, fewer days of review, and lower risk of toxicity to the thyroid gland. The PET scan measures the metabolic activity through the absorption of 2-(fluorine-18)-fluoro-2-deoxy-D-glucose (18F-FDG), which is directly proportional to the amount of disease and cell proliferation. A recent study showed that the PET and MIBG have relative sensitivity for neuroblastoma tumors, with 18F-FDG PET suggesting benefit in lower stage patients. 123I-MIBG continues to be the gold standard for imaging in all patients, including advanced stage [36].
A disadvantage of the PET is its inability to visualize lesions in the cranial vault and may have lower sensitivity for bony lesions in comparison to MIBG scans [36]. PET scan can help especially in the management and diagnosis of MIBG-negative neuroblastoma.
Alternate Imaging
In the absence of advanced technologies for radiographic imaging, it would be advisable to obtain a Tc-99 bone scan and/or skeletal survey (if Tc-99 bone scan is unavailable).
Pathology and Molecular Evaluations
The biopsy specimen must be handled with conditions that allow for relative sterility. The specimen should be fresh and not fixed in formalin. Genetic and molecular studies require at least 100 mg tissue. Specimens can be divided into following manner for appropriate diagnostic evaluations:
1.
Ten imprints without fixing with remainder fixed in formalin.
2.
Specimen in sterile culture medium, for DNA cytometry and cytogenetic.
3.
Three and four specimens are frozen and kept in liquid N2.
Fixed specimens can be evaluated for a structural study. A trained pathologist with experience in childhood solid tumors is critical to appropriate processing of tissue and selection of critical diagnostic immunohistochemical testing on the tumor specimen. Histological classification using the International Neuroblastoma Pathology Committee (INPC) guidelines will help guide risk stratification, using tumor category, MKI, and degree of differentiation [37]. Distinguishing neuroblastoma tumors from other small round blue cell tumors can be made using immunohistochemistry if the urine catecholamine is negative or unavailable [38].
Pathology report should include the following data:
Histologic category.
Histologic subtype.
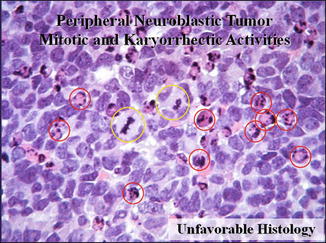
MKI in the category of neuroblastoma (Fig. 23.3).
Other data of interest such as the presence of calcification.
Potential involvement of lymph nodes and adjacent organs.
Results of the special studies.
Determination of the prognostic group according to the INPC histology favorable or unfavorable [39].
Fig. 23.3
The mitosis–karyorrhexis index (MKI) is defined as the number of tumor cells in mitosis and in the process of karyorrhexis. The index is defined this way so that mitotic activity and karyorrhexis can be assessed in relation to the cellularity of the tumor, rather than in relation to the number of high-power microscopic fields (HPFs)
The histological data obtained in a metastasis should correlate with the primary tumor and should be indicated in the report. On the contrary, after chemo treatments and/or radiotherapy histology varies greatly from area to area and there are frequent areas of necrosis and calcification; this is not relevant for prognostic assessment. Occasionally, the diagnosis of neuroblastoma may be challenging on a simple H&E staining, especially for the undifferentiated histology. The differential diagnosis should include lymphomas and leukemias, soft tissue sarcomas, and PNET/Ewing. Immunohistochemistry staining can be utilized to help guide appropriate diagnosis and management [38].
Cytogenetic and Molecular Studies
MYC-N amplification analysis: A fresh tumor tissue is required to determine MYCN amplification or chromosomal aberrations. Detection is based on fluorescence in situ hybridization (FISH) testing, real-time quantitative PCR assay, or a southern blot (Fig. 23.4).
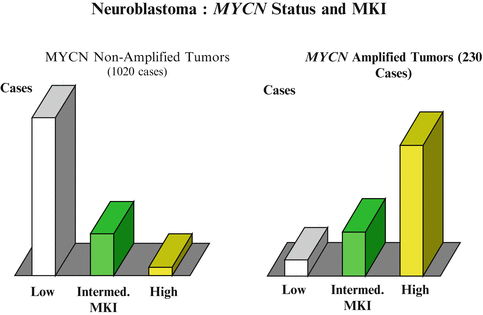
Fig. 23.4
The definition of the MYCN amplification has been defined as greater than fourfold increase in MYCN signals as compared to reference signals. MYCN amplification is found in a 30–40 % of stages 3 and 4 of neuroblastoma and in only 5 % of the stages 1, 2, or 4S neuroblastoma
Tumor cell ploidy analysis: A flow cytometric analysis on fresh tumor cell suspension is required to assess DNA index. DNA index, when available, is a useful marker of prognostic significance in infants and toddlers, with loss of utility in older children with advance disease [40].
Other Molecular Analyses
In neuroblastoma, several nonrandom genomic alterations have been described to be closely associated with distinct phenotypes of the disease [41]. Recently, loss of 11q has been reported to be highly correlated with an adverse outcome [20, 42] and has thus been proposed as a stratifying prognostic marker in the INRG classification system [15]. As 11q CNA and MYCN amplification are inversely correlated in neuroblastoma, these two genomic alterations have been suggested to delineate molecularly distinct subgroups [43, 44]. In contrast, the majority of favorable neuroblastomas lack structural genomic aberrations but show numerical variations of whole chromosomes [45, 46]. In neuroblastoma, loss of large genomic regions at 11q occurs in approximately 30 % of the tumors and is associated with an unfavorable clinical outcome [45].
Disease Risk Assignment
European and North American cooperative groups have selected various factors to define the risk in patients with neuroblastoma, but these have not been uniform, for example: The International Society of Pediatric Oncology Europe Neuroblastoma Group (SIOPEN) uses age, image-defined surgical risk factor (IDRF), and MYCN status for risk assignment in local/regional tumors, on the other hand, the Children’s Oncology Group (COG) uses age, postsurgical stage, MYCN status, DNA ploidy, loss of heterozygosity at 11q, 1p, and histology [1, 47]. Estimated 5-year survival rates for patients with non-high-risk and high-risk neuroblastoma are 90 and 50 %, respectively. Recent clinical trials have shown excellent outcomes with reduced therapy for non-high-risk disease. For patients with high-risk neuroblastoma treated with chemo radiotherapy, surgery, and stem cell transplantation, the addition of anti-disialoganglioside GD2 immunotherapy plus cytokines improves survival. Upcoming trials will study the incorporation of targeted radionuclide therapy prior to myeloablative chemotherapy into high-risk treatment [48].
A new staging system (INRG) was developed for neuroblastoma risk assignment and stratification of patients at the time of diagnosis and prior to initiating treatment (Table 23.1). This staging incorporates the extension of local, regional disease as the presence or absence of image-defined presurgical risk in L1 and L2 tumors; disseminated disease is categorized as M stage, with the exception of Ms stage, which defines the presence of metastatic disease limited to the skin, liver, bone marrow without commitment to the cortical bone, in children between the ages of 0–18 months with small primary tumors. Additional evaluations that have been recommended are molecular diagnostic tests in the tumor tissue, bone marrow analysis, and radiographic imaging [15] (Table 23.2.). Because the treatment effect of tumor excision is an inherent part of the INSS, the prognostic value of specific stages within INRGSS and INSS cannot be directly compared. The INRGSS differs from INSS in four important ways. First, it is based on preoperative imaging and image-defined risk factors (IDRFs), not surgicopathologic findings. Second, the midline is not included in the staging criteria of the INRGSS. Third, lymph node status is not included in the staging of localized disease. Fourth, whereas INSS stage 4S has an upper age limit of 12 months, the Task Force decided to extend the age group for stage MS to patients younger than 18 months. The INRGSS is a preoperative staging system that has been developed specifically for the INRG classification system. The extent of disease is determined by the presence or absence of IDRFs and/or metastatic tumor at the time of diagnosis, before any treatment. Use of this pretreatment staging system and the INRG classification system will facilitate the ability to compare results of risk-based clinical trials conducted in different regions of the world, and thereby, provide insight into optimal treatment strategies for patients with neuroblastic tumors [49].
Table 23.1
International Neuroblastoma Risk Group (INRG) Staging System (INRGSS)
INRG | Age (month) | Histologic category | Degree of differentiation of the tumor | N-MYC | Aberration of the 11q | Ploidy | Pretreatment risk group |
|---|---|---|---|---|---|---|---|
L1/L2 | Mature GN or mix GN | A very low | |||||
L1 | Any with exception mature GN or mix GNB | N/A | B very low | ||||
Amp | K High | ||||||
L2 | <18 | Anyone except mature GN or mixed GNB | N/A | No | D Low | ||
Yes | G Intermediate | ||||||
>18 | Nodular GN | Differentiated | N/A | No | E Low | ||
Neuroblastoma | Yes | ||||||
N/A | H Intermediate | ||||||
Poorly differentiated or undifferentiated | Amp | N High | |||||
M | <18 | N/A | Hyperploidy | F Low | |||
<12 months | N/A | Ploidy | I Intermediate | ||||
12 months <18 | N/A | Ploidy | J Intermediate | ||||
<18 | Amp | O High | |||||
<18 | P High | ||||||
MS | <18 | N/A | No | C Very low | |||
Yes | Q High | ||||||
Amp | R High | ||||||
Table 23.2
Descriptions of new INRG tumor stages
Tumor stage | Description |
|---|---|
L1 | Localized tumor not involving vital structures, as defined by the list of IDRFs, and confined to one body compartment |
L2 | Local-regional tumor with presence of one or more IDRFs |
M | Distant metastatic disease (except stage MS tumor) |
MS | Metastatic disease in children younger than 18 months, with metastases confined to skin, liver, and/or bone morrow |
In the resource-limited countries (RLC), there are significant limitations in the diagnosis of neuroblastoma. The majority of the centers in these countries do not have the resources to obtain all the biological and genetic data that is suggested for a complete treatment stratification and therapy assignment. Therefore, a modified stratification is typically considered based on resource availability with current use of the INSS system.
Prognostic Considerations
The Age of Presentation
Analysis of the COG and INRG data shows that age has a strong predictive value as a variable for the risk stratification. The effect of age at the time of diagnosis in the 5-year survival is profound; patients with 4S disease (7–10 % of neuroblastoma) have a more favorable outcome than other patients with metastatic neuroblastoma, often demonstrating spontaneous maturation and regression. Estimated survival rates of 70–90 % have been reported, and these tumors are usually associated with favorable biologic features. Recently, the INRG developed consensus guidelines for a modified staging system based on clinical and radiologic criteria. The INRG increased the upper limit of age from 12 to 18 months for 4S disease (now designated Ms), defined as metastases limited to skin, liver, and bone marrow (<10 %) without cortical bone involvement and either L1 or L2 stage tumors, including large unresectable primary tumors that cross the midline [50]. Children with ages from 1 to 4 years old has shown an OS >68 %, ages 5–9 years old is 52 %, and 10–14 years old is 66 % [23, 47].
Histological Classification
The INPC involves the evaluation of tumor specimens obtained prior to the treatment with respect to the degree of development and maturation, the presence of neuroblastic stroma, and the rate of mitosis-karyorrhexis [31]. These histological parameters are used to define favorable and unfavorable characteristics. Several studies have confirmed the prognostic significance of this classification system and other related systems that employ similar strategies [51] (Table 23.3).
Table 23.3
International Neuroblastoma Pathology Classification
Category and subtype | Prognostic group |
|---|---|
Neuroblastoma (Schwannian stroma-poor) | |
Undifferentiated, poorly differentiated | Differentiating FH and UH subgroups, based on the combination of age, grade of neuroblastic differentiation, and MKI class |
Ganglioneuroblastoma, intermixed (Schwannian stroma-rich) | FH |
Ganglioneuroma (Schwannian stroma-dominant) | FH |
Maturing | |
Mature | |
Ganglioneuroblastoma, nodular (Schwannian stroma-rich/stroma-dominant and stroma-poor) | UHa |
MYC-N amplification: Numerous studies have shown that MYC-N amplification is the most critical assessment in guiding treatment strategies and outcomes. This impact is most significantly noted in guiding treatment for all stage 3 and 4 and 45 NB patients less than 18 months, [51]. Children >18 months with stage 4 NB would generally be treated based on high-risk protocol and therefore less critical for therapeutic guidance [64].
Tumor cell ploidy: DNA index has been shown to be a strong predictor of outcome in infants and toddlers with metastatic disease and those with stage 4S disease [47]. The prognostic impact in other patients is less apparent in a multivariate analysis.
Additional Serum Markers: LDH, Ferritin, Neuron-Specific Enolase
Ferritin, neuron-specific enolase (NSE), and lactate dehydrogenase (LDH), are commonly assessed in children suspected to have neuroblastoma. The univariate prognostic benefit that has been suggested in numerous studies has been less obvious in multivariate analysis of large cohorts, especially in the setting of established independent prognostic factors such as age, MYC-N amplification, and stage of disease, histology, and cell ploidy.
LDH values above 1,500 U/dL in stage 4 patients were associated with a worse outcome, independent of age and stage. LDH levels >1,500 U/dL also suggested correlation with MYCN amplification, and therefore, may serve as a useful surrogate for MYCN amplification. This relationship has yet to be evaluated in a clinical trial. Similarly, NSE levels >200 ng/mL positively associated with a worse outcome only in stage 4 patients without MYCN amplification, thus limiting their utility in patient risk stratification. The ferritin >142 ng/mL has also been shown to have prognostic implications [52]. In resources-limited countries, it is therefore imperative to continue to utilize the previously accepted standard risk classification based on INSS, until advanced diagnostic resources are readily available.
Staging
International Neuroblastoma Staging System (INSS): International criteria for a common neuroblastoma staging system were first described in 1988 and subsequently revised in 1993. The INSS is a surgicopathologic staging system that depends on the completeness of resection of the primary tumor, assessment of ipsilateral and contralateral lymph nodes, and the relation of a primary tumor to the midline (Table 23.4). Although INSS has been shown to have prognostic relevance, there have been some difficulties with its widespread use. The expertise and aggressiveness of the surgeon max influence tumor staging, and lymph node sampling, extent of disease resection [53]. The INSS combines certain features of the previously used POG and CCG systems and has identified distinct prognostic groups. Based on preliminary experience, there was a need for modifications and clarifications in the INSS and International Neuroblastoma Response Criteria (INRC). A meeting was held to review experience with the INSS and INRC and to revise or clarify the language and intent of the originally proposed criteria. Substantial changes included a redefinition of the midline, restrictions on age and bone marrow involvement for stage 4S, and the recommendation that MIBG scanning be implemented for evaluating the extent of disease. Other modifications and clarifications of the INSS and INRC are presented. In addition, the criteria for the diagnosis of neuroblastoma were modified. Finally, proposals were made for the development of risk groups that incorporate both clinical and biologic features in the prediction of prognosis. The biologic features that were deemed important to evaluate prospectively included serum ferritin, NSE, and lactic dehydrogenase (LDH); tumor histology; tumor-cell DNA content; assessment of MYCN copy number; assessment of 1p deletion by cytogenetic or molecular methods [31].
Table 23.4




International Neuroblastoma Staging System
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
