Neoplasms of the thyroid
Steven I. Sherman, MD  Maria E. Cabanillas, MD
Maria E. Cabanillas, MD  Stephen Y. Lai, MD, PhD
Stephen Y. Lai, MD, PhD
Overview
Thyroid carcinomas, particularly, papillary cancers, exhibit increasing incidence but, generally, low risk for mortality. An optimal approach to an undiagnosed thyroid mass relies upon ultrasound characterization and guided fine needle aspiration of lesions at risk for clinically significant malignancy. Given the absence of prospective trials, controversy continues over the most appropriate initial treatment strategy for differentiated carcinoma, including extent of thyroidectomy and neck dissection and use of adjuvant radioiodine. Medullary carcinoma is treated initially with thyroidectomy, central and possible lateral neck dissections, and early evaluation to distinguish sporadic from hereditary disease. Recent developments in systemic, targeted therapies have led to multiple anti-angiogenic drugs available to treat progressive metastatic differentiated or medullary carcinoma with significant improvement in progression-free survival, though improvements in overall survival are lacking. In distinct contrast, anaplastic carcinoma remains one of the most fulminantly aggressive malignancies, with limited and generally palliative benefit only from selected use of multimodality therapy with surgery, radiation, and chemotherapy.
Historical perspective
Thyroid carcinoma, historically an uncommon disease, has transformed into one of the most commonly diagnosed malignancies, with escalating worldwide incidence. Although most patients require localized therapy only, treatment of advanced disease has changed with advent of targeted therapies. Even initial treatment has undergone revision, with recognition that more conservative approaches are sufficient.
Incidence and epidemiology—local and worldwide
About 62,450 persons were diagnosed in 2015 with carcinoma of the thyroid gland in the United States, with disease-related mortality of nearly 2000.1 Globally, the incidence is six times higher, whereas disease-related mortality is >20 times higher.2 Female incidence is about twice that of males, peaking in South Korea, where thyroid cancer is the leading malignancy diagnosed in women. The escalating incidence is seen primarily in small tumors confined to the gland and has been associated with increased thyroid imaging, higher socioeconomic status, and “overdiagnosis.”3
Differentiated thyroid carcinomas (DTC), derived from the hormone-producing follicular epithelial cells, account for 96% of thyroid cancers, including papillary thyroid cancer (PTC), follicular thyroid cancer (FTC), and oxyphilic carcinomas (88%, 6%, and 2%, respectively).4 Another 2% are medullary thyroid cancer (MTC) carcinomas, derived from neuroendocrine “C” cells, and the remaining 1% are poorly differentiated thyroid cancer (PDTC) carcinoma or highly aggressive, undifferentiated, anaplastic thyroid cancer (ATC) carcinomas.
Risk factors—genetic, behavioral, environmental
Although most lack an obvious cause for DTC, the best established risk factor is radiation exposure, especially during childhood and adolescence. Between 0.1 and 10 Gy of exposure, the excess relative risk is 7.7 per Gy, with risk leveling above 10 Gy and after age 20 at time of exposure.5 Obesity has also been associated with higher risk of DTC (relative risk about 1.3).6 Iodine deficiency has been associated with a higher frequency of FTC, and possibly contributes to risk for ATC.7 DTC is a component of certain genetic syndromes, including familial adenomatous polyposis, Cowden syndrome, and Carney complex.8, 9 Familial nonmedullary carcinoma, occurring in at least two first-degree relatives, has been reported in 5% of all PTC patients, although genetic loci and mechanisms are unclear.10 Hereditary MTC syndromes, including subtypes of multiple endocrine neoplasia (MEN) type 2A and MEN2B, account for 25% of all cases of MTC; environmental factors are not known to influence development of MTC.11
Prevention
Other than use of potassium iodide prophylaxis in the setting of a nuclear accident or mechanical shielding of the thyroid from external radiation,12 there is no known strategy to prevent development of DTC. Thyroidectomy following prospective screening of children at high risk for hereditary MTC may prevent development of aggressive malignancy.13
Pathology
Conventional PTCs are characterized by papillae with a fibrovascular core surrounded by tumor cells with large, oval, often overlapping nuclei containing hypodense powdery chromatin, cytoplasmic pseudoinclusions, and grooves (Figure 1). The follicular variant accounts for about 10% of PTCs, with cells displaying nuclear features of PTC but organized into follicles rather than papillae. In contrast, the uncommon tall cell variant of PTC is a more aggressive tumor, characterized by eosinophilic tumor cells that are twice as tall as they are wide. The primary tumors are larger, invasive, and frequently metastatic.
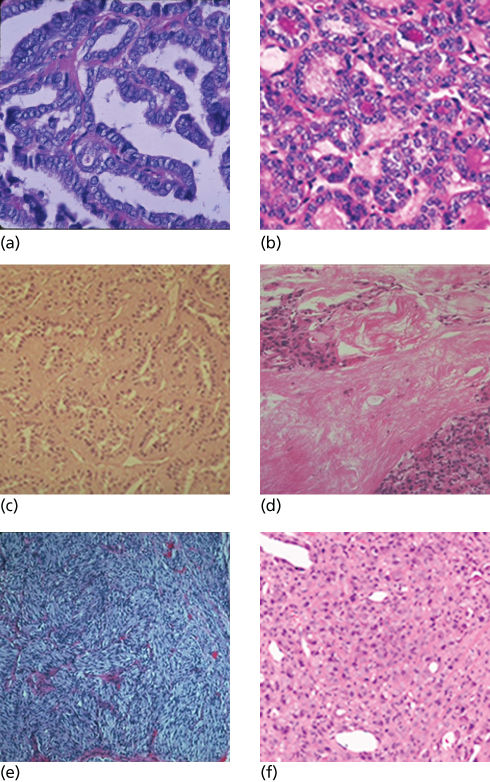
Figure 1 Pathologic features of thyroid carcinoma. (a) Papillary carcinoma, with characteristic fibrovascular papillary formation, crowded nuclei, and nuclear clearing. (b) Follicular variant of papillary carcinoma, with typical cells of papillary carcinoma in follicular formations. (c) Tall cell variant of papillary carcinoma, with cells at least twice as tall as they are wide and eosinophilic cytoplasm. (d) Follicular carcinoma, demonstrating invasion across a thick capsule into neighboring thyroid. (e) Medullary thyroid carcinoma, with nests of spindle-shaped cells. Such nests are often interspersed with clusters of round-to-oval cells. (f) Anaplastic carcinoma, showing large, discohesive, pleomorphic tumor cells.
Source: Courtesy of Michelle D. Williams M.D.
FTCs are distinguished from follicular adenomas by demonstrating invasiveness in one or more foci along the capsule or across vascular endothelial walls. The extent of invasion separates FTC into minimally invasive and widely invasive lesions, the former demonstrating only scattered foci of capsular or vascular invasion. Cytologic features do not reliably distinguish benign from malignant follicular lesions. Oxyphilic (or Hürthle cell) neoplasms are formed by cells containing numerous mitochondria, imparting a granular, eosinophilic cytoplasmic appearance.14 Most have a follicular architecture and are diagnosed as carcinomas by the presence of invasion.
PDTC is often considered intermediate between DTC and ATC and may represent a partial dedifferentiated state. Cells tend to be nested with solid or insular architectural patterns, without the nuclear features of PTC. Diagnostic criteria also require one of (1) convoluted nuclei, (2) tumor necrosis, or (3) at least three mitoses per high power field.15 ATC is usually macroscopically infiltrative; microscopically, the tumor is heterogeneous, with pleomorphic nuclei, sheets of spindle, epithelioid, or giant cells, areas of necrosis, and extensive inflammatory infiltrate. While PTC, FTC, and even PDTC generally stain positive for thyroglobulin (Tg) and other follicular cell-specific antigens, ATC may stain only for cytokeratin and PAX8.16
MTCs commonly contain nests and sheets of rounded or spindle-shaped neuroendocrine cells, surrounded by a fibrovascular stroma. Nuclei tend to have speckled chromatin, and the cytoplasm is usually granular and eosinophilic. Amyloid deposition within the stroma is pathognomonic. Calcitonin and neuroendocrine immunohistochemical markers are usually detected. MTC in hereditary syndromes arises from a background of C cell hyperplasia.
Pathogenesis and natural history
Molecular alterations
Oncogenes associated with PTC include mutually exclusive activating mutations in BRAF (62%), RAS (13%), and RET/PTC chromosomal rearrangements (7%), all upstream of the mitogen-activated protein (MAP) kinase.17 BRAF mutations are most common in conventional PTC and tall cell variants, whereas RAS mutations are seen in the follicular variant of PTC and FTC. Comprehensive analysis of genomic, transcriptional, epigenetic, and microRNA changes identifies clear signaling differences between BRAF and RAS-type tumors.17 In FTC, chromosomal rearrangements fusing a thyroid-specific transcription factor to peroxisome proliferator-activated receptor gamma-1 to form the PAX8-PPARγ oncogene may also be seen.18 In PDTC and ATC (which may arise from pre-existing DTC), coexisting mutations are more common, including RAS, BRAF, TP53, CTNNB1, PIK3CA, and PTEN mutations and loss of heterozygosity. Other factors contributing to progression of DTC include overexpression of other intracellular kinases, deoxyribonucleic acid hypermethylation and histone deacetylation leading to silencing of tumor-suppressor genes, and cell-cycle dysregulation.17, 19
Mutations in exons 8, 10, 11, and 13–16 of the tyrosine kinase receptor RET occur in nearly all families with hereditary MTC.10, 11 Somatic mutations are also found in 40–50% of sporadic MTC tumors, particularly the highly activating codon 918 mutation associated with poorer patient prognosis.20 Strong genotype–phenotype correlations allow stratification of mutations to predict disease aggressiveness.21 RAS mutations are reported in 20–70% of sporadic MTC lacking RET changes.
Screening
RET mutations in hereditary MTC are inherited in an autosomal dominant manner with generally high penetrance. In addition to known kindreds, about 6% of patients with clinically sporadic MTC carry a germ line mutation in RET, leading to identification of new families.22 Genetic counseling and testing for RET proto-oncogene mutations should be offered to all patients newly diagnosed with clinically apparent sporadic MTC, as well as for screening children and adults in known families with inherited forms of MTC.21, 23 In the patient with clinically sporadic MTC, the likelihood of a false negative RET germ line test for familial disease is <1%; however, if the family history is suggestive for an autosomal inherited disorder, complete sequencing of RET can be considered, and the family should be screened clinically and biochemically for possible MTC. For patients with familial adenomatous polyposis or Cowden syndrome, at higher risk for DTC, annual examination and possible ultrasound of the thyroid have been suggested.24
Diagnosis
While about 5% of individuals have a palpable thyroid nodule, and ultrasonography reveals nodules in up to half of individuals without palpable lesions, only 5% of nodules are malignant.25, 26 Most are asymptomatic, and few clinical features are specific for malignancy. Sonographic criteria that increase malignancy likelihood include microcalcifications, irregular margins, and shape taller than wide.27 Incidentally detected thyroid nodules are also seen in 1–2% of fluorodeoxyglucose-positron emission tomography (FDG-PET) scans, for which the risk of malignancy is 40%.28 Thyroid ultrasound with cytologic examination of a fine needle aspirate (FNA) of solitary nodules at least 1 cm in size is the most appropriate diagnostic procedure; in the setting of multiple nodules, those with suspicious sonographic appearances (and if none are present, the largest) should be preferentially aspirated.29 PTC, MTC, and ATC can be readily diagnosed on the basis of cytologic criteria.30 The false-positive and false-negative rates for nodules characterized as “malignant” and “benign,” respectively, are <5%. However, as FTC is distinguished from benign follicular adenoma by invasion through the tumor capsule or vascular wall, these nodules are usually reported cytologically (along with follicular variants of PTC) as indeterminate follicular lesions or neoplasms, with an overall 20% risk of malignancy.30, 31 Molecular testing with a gene expression classifier can reduce the risk of malignancy in indeterminate nodules to as low as 5%; detection of BRAF mutations, although uncommon in indeterminate nodules, carries a very high positive predictive value for malignancy.32–35 Although not routinely recommended for initial nodule evaluation, a random serum calcitonin level >100 pg/mL has high predictive value for MTC. Once malignancy is suspected, ultrasonography of the nodal chains in the neck is recommended before surgery; computed tomography (CT) or magnetic resonance imaging (MRI) may also provide occasional benefit in surgical planning for invasive or bulky disease.
Patients with aggressive disease, especially PDTC and ATC, commonly present with extensive local invasion and distant metastases.36 The lungs and pleura are the most common sites of distant metastases, being seen in up to 90% of patients with distant disease, followed by bones, brain, skin, liver, and kidneys. CT of the neck and mediastinum can accurately determine the extent of the thyroid tumor and identify tumor invasion of the great vessels and upper aerodigestive tract structures. Metastatic work-up to detect distant disease is necessary but should not delay therapy.37
In half of MTC patients, metastatic cervical adenopathy is noted at initial presentation, and symptoms of upper aerodigestive tract compression or invasion are reported in up to 15% of patients with sporadic disease. Distant metastases are most commonly found in the liver. The ability of the tumor to over-secrete measurable quantities of Ct, occasionally along with other peptides and biogenic amines such as adrenocorticotrophic hormone or calcitonin gene-related peptide, leads to unexplained diarrhea or symptoms of Cushing syndrome in many patients.
TNM staging, classification
The seventh edition of the American Joint Committee on Cancer (AJCC)/UICC (Union for International Cancer Control) staging system is recommended for standard clinicopathologic staging and assessment of risk for mortality for all thyroid cancers (Table 1).21, 37, 38, 40 However, there are significant concerns that it may overestimate risk for small tumors and underestimate risk for young patients with DTC.41, 42 The important prognostic value of histologic subtypes of DTC is also excluded. Risk for DTC recurrence may be better predicted by other proposed schemes following completion of initial therapy.40, 43, 44
Table 1 Tumor, node, metastases (TNM): The American Joint Committee on Cancer (AJCC) Staging Scheme for Thyroid Carcinomas (Seventh Edition).38
| T: Tumor status | ||
| T0 | No evidence of primary tumor | |
| T1a | Tumor ≤1 cm, without extrathyroidal extension | |
| T1b | Tumor >1 cm but ≤2 cm in greatest dimension, without extrathyroidal extension | |
| T2 | Tumor >2 cm but ≤4 cm in greatest dimension, without extrathyroidal extension | |
| T3 | Tumor >4 cm in greatest dimension limited to the thyroid -or- Any size tumor with minimal extrathyroid extension (e.g., extension into sternothyroid muscle or perithyroidal soft tissues) | |
| T4a | Tumor of any size extending beyond the thyroid capsule to invade subcutaneous soft tissues, larynx, trachea, esophagus, or recurrent laryngeal nerve | |
| T4b | Tumor of any size invading prevertebral fascia or encasing carotid artery or mediastinal vessels | |
| Tx | Tumor status unknown | |
| N: Regional node status | ||
| N0 | No metastatic nodes | |
| N1a | Metastases to level VI (pretracheal, paratracheal, and prelaryngeal/Delphian lymph nodes) | |
| N1b | Metastases to unilateral, bilateral, or contralateral cervical (level I, II, III, IV, or V) or retropharyngeal or superior mediastinal lymph nodes (level VII) | |
| Nx | Nodes status unknown | |
| M: Distant metastases | ||
| M0 | No distant metastases | |
| M1 | Distant metastases | |
| Mx | Metastases status unknown | |
| <45 years | ≥45 years | |
| Stage assignments: differentiated thyroid carcinoma | ||
| Stage I | Any T, any N, M0 | T1, N0 M0 |
| Stage II | Any T, any N, M1 | T2, N0, M0 |
| Stage III | T3, N0, M0 | |
| T1–3, N1a, M0 | ||
| Stage IV | A: T4a, any N, M0 | |
| Any T, N1b, M0 | ||
| B: T4b, any N, M0 | ||
| C: Any T, any N, M1 | ||
| Stage assignments: medullary thyroid carcinoma | ||
| Stage I | T1, N0, M0 | |
| Stage II | T2–3, N0, M0 | |
| Stage III | T1–3, N1a, M0 | |
| Stage IVA | T4a, N0, M0 | |
| Stage IVB | T4a, N1a, M0 | |
| Any T, N1b, M0 | ||
| Stage IVC | Any T, any N, M1 | |
| Anaplastic thyroid carcinoma | ||
| All are classified as stage IV | ||
Source: Edge 2010. Reproduced with permission of Springer.
Prognostic factors
In the Surveillance, Epidemiology, and End Results (SEER) report of 15,700 patients, the overall 10-year age- and gender-corrected survival rates were 98% for PTC, 92% for FTC, 80% for MTC, and 13% for ATC.45 Older age at diagnosis and greater extent of disease including increasing tumor size, extrathyroidal invasion and distant metastases, are associated with a worse prognosis, independent of the type of cancer. In PTC, BRAF mutations may be prognostic for locoregional recurrence and perhaps mortality, whereas in MTC, RET M918T mutations are associated with worse outcomes.20, 46–48
Multidisciplinary care
Surgery—standard operations, complications, outcomes
Total thyroidectomy has been the initial surgical procedure advocated for most patients with DTC, given that (1) foci of PTC are commonly bilateral49, 50; (2) contralateral recurrence occurs in 5–10% of patients who undergo unilateral surgery; and (3) efficacy of therapy with radioiodine is maximized by total resection. Earlier cohort studies support the contention that recurrence rates are 2–3 times higher following lobectomy compared with total thyroidectomy for primary PTC >1 cm, with possible improvements in survival in multivariate analyses.51–53 More recently, larger studies have challenged these conclusions, suggesting no improvement in survival following total thyroidectomy.54, 55 Active surveillance has even been advocated for selected patients with small PTC confined to the gland, given infrequent progression on serial imaging.56 In the absence of randomized prospective trials, total thyroidectomy is recommended for DTC patients with primary tumors >4 cm or clinical evidence preoperatively of extrathyroidal invasion or nodal or distant metastases. Either total thyroidectomy or ipsilateral lobectomy may be appropriate for smaller primary tumors without metastatic or invasive disease and lobectomy or active surveillance optimal for subcentimeter primary tumors.
Although microscopic regional nodes with PTC occur in up to 80% of patients, only about 35% have clinical cervical or mediastinal node metastasis, typically in lateral cervical compartments.57 With clinically involved lateral neck disease, a comprehensive neck dissection (levels II–V) should be performed.58, 59 While resection of clinically involved central compartment (level VI and superior mediastinal nodes) nodes has been associated with improved survival and is commonly recommended, the potential benefit of prophylactic central neck dissection may outweigh the potential surgical morbidity only when performed by experienced thyroid surgeons.60 In the presence of invasion of aerodigestive tract structures, similar survival rates are achieved from either complete surgical resection or shave excision leaving only microscopic residual disease.61 However, half of patients die within 4 years following shave excision for frank cartilage destruction or intraluminal involvement. Surgery in patients with extensively invasive thyroid carcinoma should aim, therefore, to remove all gross tumors, maximizing functionality, unless the disease is unresectable or patient refuses the proposed surgery.62
For newly diagnosed MTC, serum calcium and plasma-free metanephrines should be measured to exclude coexistent hyperparathyroidism and pheochromocytoma, unless MEN2 has been ruled out by germ line RET testing. Total thyroidectomy is indicated in all patients with MTC, especially given the high frequency of bilateral disease in both sporadic and familial disease, and has been associated with improved survival.21, 63 Even in the absence of clinically detectable nodal metastases, central neck compartment dissection should be performed in all patients, and ipsilateral lateral neck and/or mediastinal dissections should be strongly considered when the primary tumor is >1 cm or when central compartment disease is present. Disfiguring radical neck dissections do not improve prognosis and are not indicated. In the presence of grossly invasive disease, more extended procedures with resection of involved neck structures may be appropriate, but function-preserving approaches are preferred.
Given the identification of patients with malignant disease as early as age 6 years, prophylactic thyroidectomy by age 5 is recommended for carriers of a familial RET
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


