COMPARABLE ASPECTS
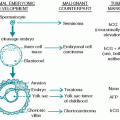
The World Health Organization (WHO) classification of the chronic myeloproliferative disorders (MPDs) includes polycythemia vera (PV), chronic idiopathic myelofibrosis (MF), essential thrombocythemia (ET), chronic eosinophilic leukemia (CEL)/hypereosinophilic syndrome (HES), chronic myelogenous leukemia (CML), chronic neutrophilic leukemia, and unclassifiable chronic MPD. Chronic myelomonocytic leukemia (CMML) has features of both an MPD and a myelodysplastic syndrome (MDS). Details on CML and CMML are presented in
Chapter 23. This chapter focuses on PV, ET, MF, CEL/HES, and systemic mastocytosis (SM).
The MPDs each result from a genetic alteration within a pluripotent hematopoietic progenitor cell that induces the excessive production of one or more cell lineages. The individual diseases are distinguished by the predominant lineage that is overproduced.
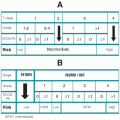
Table 24.1 compares important clinical and distinguishing features of the MPDs. There is considerable overlap between several of the MPDs. Long-term observation may be required to clarify the diagnosis. Unclassifiable MPDs is the best designation for patients who have leukoerythroblastic blood smears, normal red blood cell mass, and a hypercellular marrow that shows only mild fibrosis.
Erythrocytosis, granulocytosis, eosinophilia, basophilia, and thrombocytosis may be due to disorders other than MPDs, as discussed in
Chapter 34, Sections I to V in “Increased Blood Cell Counts.” Similarly, bone marrow fibrosis may be secondary to a variety of other etiologies, as discussed in
Chapter 34, Section I.B in “Cytopenia.”
I. PATHOGENESIS. The MPDs are clonal neoplastic disorders that arise from a single pluripotential hematopoietic stem cell. The molecular abnormalities underlying many of the MPDs are rapidly becoming elucidated.
A. Molecular and cytogenetic abnormalities. A mutation of the Janus kinase 2 (JAK2) gene has been identified in PV, ET, and MF. The JAK2 protein is a tyrosine kinase that is phosphorylated by the receptors for erythropoietin (EPO), thrombopoietin, granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, and interleukin-3 in response to ligand binding. Activation of JAK2 in this way initiates a signaling cascade that induces cell proliferation in response to these growth factors.
1. The most commonly observed mutation results in the replacement of valine by phenylalanine at position 617 (V617F) in exon 14 of JAK2. The mutated protein enables hematopoietic cells to survive in the absence of growth factors and to have enhanced proliferation when exposed to low growth factor concentrations.
The JAK2 V617F mutation occurs in approximately 95% of PV, 50% of ET or MF, 20% of unclassifiable MPD, and 2% of HES. The mutation is homozygous in approximately 40% of PV due to mitotic recombination. Mutations involving exon 12 of JAK2 have been identified in about 4% PV patients.
In addition, point mutations at position 515 (exon 10) of the thrombopoietin receptor (MPL) have been identified in 5% to 10% of patients with
MF or ET. The mutated MPL receptor activates the
JAK signaling pathway in the absence of thrombopoietin.
2. Mutations have been identified in additional genes that encode proteins involved in critical cell processes.
a. Kinase signaling. CBL ubiquitinates receptor tyrosine kinases, leading to their inactivation. Loss-of-function CBL mutations have been observed in MF. LNK inhibits JAK2 signaling. Inactivating mutations of LNK have been reported in ET and MF.
b. Epigenetic regulation. TET2 transfers a hydroxyl group to methylcytosine of DNA. Mutations of TET2 have been reported in 5% to 20% of MPDs. ASXL1 participates in histone demethylation and is mutated in <10% of MPD patients. EZH2 plays a role in histone methylation and is mutated in approximately 13% of MF cases.
3. Chromosome analyses have established that a clonal cytogenetic abnormality is present in erythroblasts, neutrophils, basophils, macrophages, megakaryocytes, and subsets of B lymphocytes, but not in fibroblasts. Abnormal karyotypes are found in about 20% of PV cases at the time of diagnosis, with deletions of 20q or 13q or trisomies of 8 or 9 being most common.
4. In MF, abnormal karyotypes are found in 35% of cases; deletions of 20q or 13q and partial trisomy 1q account for 70% of the abnormal karyotypes found. The frequency of chromosomal abnormalities increases over time, particularly if patients are treated with chemotherapeutic agents. In some cases of CEL, a very small interstitial deletion on chromosome 4q12 fuses the FIP1L1 and platelet-derived growth factor receptor (PDGFR)A genes, producing a novel transforming fusion gene. The t(5;12)(q33;p13) occurs in other CEL cases and fuses the PDGFRB gene to the ETV6 gene.
B. Hematopoiesis in the MPDs is generally characterized by autonomous growth of progenitor cells in the absence of growth factors and hypersensitivity to the proliferative effects of growth factors.
1. Erythropoiesis in vitro in semisolid media normally requires exogenous EPO. Bone marrow progenitor cells from patients with PV form colonies in vitro without exogenous EPO and proliferate in response to very low EPO concentrations. Serum EPO levels are usually low in PV and are normal or elevated in most cases of secondary polycythemia.
2. Granulocytopoiesis is frequently increased in all MPDs to varying degrees and is manifested by neutrophilia and myeloid hyperplasia in the marrow.
3. Megakaryocytopoiesis. Megakaryocyte progenitors from ET patients are able to grow autonomously in vitro without added thrombopoietin.
4. Extramedullary hematopoiesis occurs in the liver and spleen in patients with MF and contributes to organ enlargement. However, the degree of organomegaly does not correlate well with the level of extramedullary hematopoiesis.
C. Bone marrows in MPDs demonstrate hypercellularity that is often trilineage but are diagnostic of a specific disorder only in MF. Megakaryocytes are greatly increased in number and size in ET and MF at all stages of disease and to a lesser degree in PV. Reticulin also can be increased in all MPDs, but collagen fibrosis occurs only in MF and in PV that has converted to MF.
1. Fibrosis of the marrow develops in all patients with MF and in many patients with PV or ET over time. The fibrosis is caused by the release of cytokines, including transforming growth factor-β and basic fibroblast growth factor, from clonal megakaryocytes or monocytes. The growth factors act on nonclonal fibroblasts and stromal cells and induce increased deposition of various interstitial and basement membrane glycoproteins, including collagen types
I, III, IV, and V. Type III collagen is uniformly and preferentially increased. The fine reticulin fibers that are visible with silver stains are principally type III collagen and do not stain with trichrome dyes.
2. MF. Marrow fibrosis is prominent in MF. Megakaryocytes are increased in number, and they are atypical, enlarged, and immature. Neutrophilic granulopoiesis is hyperplastic. A marked neovascularization is also present, even in the early proliferative phase of the disease.
3. PV. Trilineage hyperplasia in the marrow is the hallmark of PV. Erythroid hyperplasia is prominent. Megakaryocytes are enlarged, clustered, mature, and pleomorphic with multilobulated nuclei. Iron stores are absent or decreased in most untreated patients. In secondary erythrocytosis, erythroid hyperplasia may be present, but megakaryocytes remain small and normal with no tendency to cluster.
4. ET. Increased numbers of enlarged megakaryocytes with mature cytoplasm and multilobulated nuclei and a tendency to cluster in a bone marrow with normal or slightly increased cellularity constitute the hallmarks of ET. In reactive thrombocytosis, increased numbers of megakaryocytes may be present, but they have normal size and morphology and no tendency to cluster.
II. COMPLICATIONS OF THE MPD
A. Thrombotic phenomena, both venous and arterial, can complicate PV, ET, and MF. Myocardial ischemia and cerebrovascular ischemia are the most serious events, but thrombosis can occur anywhere in the venous or arterial system. For example, PV is the most common cause of hepatic vein thrombosis (Budd-Chiari syndrome). In PV, two-thirds of thrombotic events occur either at presentation or before diagnosis, and one-third occur during follow-up.
1. Thrombosis risks and prevention in PV and ET. Risk factors for thrombosis in PV and ET include age ≥60 years and history of a prior thrombotic event. Additional risk factors for thrombosis include elevated WBC (>15,000/µL) at presentation, hypertension, hyperlipidemia, diabetes, and smoking history. In ET and MF, the V617F mutation conveys an increased risk of thrombosis.
In PV, the risk of thrombosis increases with the hematocrit, so phlebotomy is performed to keep the hematocrit <45% in men and <42% in women. Low- and intermediate-risk (0 risk factors) PV and ET patients are given low-dose aspirin. Aspirin appears to be of most benefit in low-risk ET if the JAK2 V617F mutation is present. High-risk (two risk factors) patients also receive hydroxyurea (HU; also known as hydroxycarbamide). In ET, the level of the platelet count does not correlate with the risk of thrombosis. However, reducing the platelet count and WBC is associated with a reduced risk of thrombotic events in retrospective studies.
HU is more effective than anagrelide in preventing arterial thrombotic events in patients at high risk for thrombosis. Aspirin administration to ET patients with very high platelet counts (>1,000,000/µL) can be associated with increased bleeding risk.
B. Microvascular arterial thrombosis is easily and best controlled by low-dose aspirin or by reduction of platelet count to normal levels.
1. Erythromelalgia is the most characteristic vaso-occlusive manifestation in MPDs and is most often associated with PV or ET. It is caused by the toxic effects of platelet arachidonic acid on arterioles. Localized painful erythema and warmth occur in the distal portions of the extremities and may progress to cyanosis or necrosis of toes or fingers.
2. Microvascular arterial thrombosis in PV or ET is usually transient and not progressive. Manifestations can include ocular disturbances, amaurosis fugax, diplopia, headache, vertigo, hypesthesia, paresthesia, dysarthria, aphasia, and syncope. If superimposed on a previously compromised vasculature, these events could result in stroke, myocardial infarction, or digital gangrene.
C. Hemorrhagic phenomena occur in PV, ET, and late MF but are less common than thrombotic events. Easy bruisability and purpura are the usual manifestations. Bleeding can be treated by decreasing the platelet count, platelet transfusion, or administering desmopressin (DDAVP).
1. Bleeding can occur spontaneously without relationship to the platelet count if there is abnormal platelet function, but it occurs especially when the count exceeds 1,000,000/µL. Caution should be exercised in administering aspirin in this setting.
2. Acquired von Willebrand disease develops occasionally in the MPDs. This coagulopathy is characterized by a very high platelet count, a normal or prolonged bleeding time, normal factor VIII, and normal von Willebrand factor (vWF) antigen levels, but with decreased vWF—ristocetin cofactor activity, decreased collagen-binding activity, and a decrease or absence of large vWF multimers. This condition simulates type II vWF deficiency. A ristocetin cofactor activity should be checked, and aspirin should be used cautiously if activity is <50%.
D. Hypercatabolism. Hyperuricemia and hyperuricosuria are present in nearly all patients with active MPD. Treatment with allopurinol can prevent gouty arthritis, uric acid nephropathy, and nephrolithiasis, but its necessity is unproven. Pruritus is a frequent problem, particularly in PV. Fever, heat tolerance, and weight loss ensue when the disease becomes rapidly progressive.
E. Interconversions of the MPDs are uncommon. The only consistent transformation is the conversion of PV or ET into MF.
F. Transformation to acute myelogenous leukemia (AML). The risk of progressing to AML is approximately 2% for ET, 5% for PV, and 10% for MF within 10 years of diagnosis. The risk for AML transformation is higher in patients with MF who have undergone splenectomy. Prior treatment with alkylating agents or radioactive phosphorus (32P) also increases the risk.
G. Misleading laboratory results
1. Normal hematocrits in patients with PV can represent erythrocytosis masked by hemorrhage, iron deficiency, increased plasma volume, or splenomegaly with sequestration.
2. Pseudocoagulopathy. Prolonged clotting times in patients with marked erythrocytosis are usually the result of excessive amounts of anticoagulant relative to the small plasma volume in the test tube. Accurate determinations can be made if the volume of anticoagulant is adjusted for the hematocrit.
3. Pseudohyperkalemia. Marked thrombocytosis may result in elevated serum potassium concentrations because platelets release potassium during the clotting reaction. The true level is determined by measuring the potassium concentration in plasma rather than in serum.
4. Pseudo—hyper-acid-phosphatemia. Platelets are rich in acid phosphatase. Marked thrombocytosis may result in spurious elevations of enzyme levels measured in serum and plasma.
5. Pseudohypoglycemia. Leukocytes metabolize glucose from serum in test tubes. Dramatically low blood glucose concentrations may result from
marked granulocytosis. More accurate glucose levels can be measured if the sample is analyzed immediately after the sample is drawn.
6. Pseudohypoxemia. Oxidative respiration is used by monocytes and immature leukocytes to a greater extent than by mature leukocytes and platelets and is not used by mature erythrocytes. Falsely low oxygen tensions may be seen in patients with severe thrombocytosis or granulocytosis because of oxygen consumption within the test tubes. The presence of hypoxemia may be clarified if specimens are collected in test tubes containing fluoride and are immediately placed in ice.
POLYCYTHEMIA VERA
See “
Comparable Aspects” at the beginning of this chapter for pathogenesis, bone marrow findings, complications, and misleading laboratory results of the MPDs. Familial cases occur occasionally in PV.
I. DIAGNOSIS. PV is a clonal MPD that harbors a JAK2 mutation (most commonly V617F) in approximately 95% of cases. Therefore, mutation analysis of the JAK2 gene will help to segregate PV from secondary causes of erythrocytosis.
A. WHO criteria (2008). The diagnosis of PV requires both major criteria plus any minor criterion or the A1 criterion plus any two minor criteria.
1. Major criteria
2. Minor criteria
B. Laboratory studies
1. Red blood cell mass (RBCM). Autologous RBCs are 51Cr-labeled and injected intravenously, and then a blood sample is drawn to quantitate the dilution of the labeled cells and calculate the circulating RBCM. RBCM using 51Cr is rarely available today.
2. Clonal genetic abnormality. The presence of the JAK2 V617F or exon 12 mutation in the blood or bone marrow is adequate to demonstrate a clonal etiology for the erythrocytosis. Absence of a
JAK2 mutation suggests a secondary cause for the erythrocytosis (see “
Comparable Aspects,” Section IA).
3. Erythroid colony-forming assay. PV bone marrow demonstrates EPO independency in culture. However, this assay is cumbersome and not routinely performed by clinical laboratories.
4. Supportive studies
a. CBC. Erythrocytes are usually normocytic and normochromic unless iron deficiency is present. Poikilocytosis and anisocytosis accompany the transition into MF late in the disease course. Granulocytosis in the range of 12,000 to 25,000/µL occurs in two-thirds of patients at presentation. Early forms may be present but are not frequent. Two-thirds of patients have basophilia. Platelet counts usually are in the range of 450,000 to 800,000/µL, occasionally with abnormal morphology.
b. Serum EPO levels can be normal or reduced in PV. Although autonomous expansion of the RBCM would be expected to suppress EPO production, this assay cannot reliably distinguish between PV and EPO-driven erythrocytosis. EPO production is depressed and circulating EPO catabolism is increased as the RBCM expands from any cause. Furthermore, a normal serum EPO level is common in hypoxic erythrocytosis unless the hypoxemia is extreme.
c. Abdominal ultrasonography or CT scanning can rule out renal or hepatic causes of erythrocytosis and quantitate spleen size.
d. Bone marrow examinations can be used to demonstrate panmyelosis and abnormal megakaryocyte morphology consistent with PV, quantitate the extent of reticulin fibrosis if transition to MF is suspected, or evaluate the percentage of blasts if transformation to AML is a concern.
C. Differential diagnosis includes the other MPDs and relative or secondary erythrocytosis (see
Chapter 34, Section I in “Increased Blood Cell Counts”). Reduced plasma volume, hypoxemia (e.g., altitude, emphysema, sleep apnea), renal cysts or carcinoma, hepatic neoplasms, or uterine myomata can cause sec ondary erythrocytosis.
II. CLINICAL COURSE. The survival of patients with PV approaches that of a matched otherwise healthy population with modern therapy. The median survival exceeds 12 years.
A. Predominant signs and symptoms early in the disease are secondary to increased red blood cell mass that results in plethora and hyperviscosity. Modest splenomegaly is present in 75% of cases and hepatomegaly in 40%. Splenomegaly is caused by an increased splenic red blood cell pool and not by extramedullary hematopoiesis, which is absent early in the disease. Pruritus develops in 15% to 50% of cases, urticaria in 10%, and gout in 5% to 10%.
1. Hyperviscosity results in decreased blood flow and, consequently, in tissue hypoxia. Manifestations include headache, dizziness, vertigo, tinnitus, visual disturbances, stroke, angina pectoris, claudication, and myocardial infarction.
2. Thrombotic manifestations can result from hyperviscosity associated with the increased RBCM.
a. Types of events. Both arterial and venous thromboses occur in PV, more commonly in women than in men. Approximately two-thirds of the thrombotic events are severe and life-threatening, including cerebrovascular accidents, myocardial infarctions, pulmonary infarctions, and axillary, hepatic, portal, splenic, or mesenteric vein thromboses. The remaining one-third of events are uncomplicated deep vein or other thromboses.
3. Hemorrhagic manifestations (10% to 20% of patients) include epistaxis, ecchymosis, and gastrointestinal (GI) bleeding. Minor mucosal bleeding is most common. Acquired abnormalities of vWF can occur with marked thrombocytosis (see “
Comparable Aspects,” Section II.C).
B. Phases of disease
1. Erythrocytic phase. The phase of persistent erythrocytosis that necessitates regular phlebotomies lasts from 5 to 25 years. The manifestations of erythrocytosis and severity of complications depend on comorbid conditions and sufficient use of phlebotomy.