INTRODUCTION
SUMMARY
Myeloma derives from the proliferation of a clone of malignant plasma cells that secretes a complete and/or partial monoclonal immunoglobulin protein. It originates in most, perhaps all, cases from an antecedent monoclonal gammopathy (essential monoclonal gammopathy) that progresses by clonal evolution (acquisition of additional mutations) to a malignant B-cell malignancy, often myeloma, at a rate of 1 percent per year. Myeloma cells accumulate in the marrow microenvironment where contact with extracellular matrix and interaction with marrow accessory cells, such as osteoblasts, osteoclasts, and stromal cells, evokes cell growth and cell-survival signals, and contributes to resistance to therapy. Myeloma cells show a complex genomic phenotype, with chromosomal translocations and small copy number variations that affect patient prognosis. Patients with myeloma have signs resulting from marrow infiltration (anemia), bone destruction (bone pain, pathologic fractures), excessive immunoglobulin production and deposition (renal failure and amyloidosis-related symptoms), and immunosuppression (e.g., infection). Clinical manifestations of myeloma vary as a result of the heterogeneous biology, spanning the entire spectrum from indolent to highly aggressive disease with extramedullary features. Diagnostic workup of myeloma should include serum protein electrophoresis together with immunoglobulin immunofixation, serum-free light-chain assay, a 24-hour urine collection to quantitate urinary protein, basic blood metabolic panel including blood counts and renal function, and marrow aspirate or biopsy with fluorescence in situ hybridization and cytogenetic studies. Radiographic bone survey is used to detect osteopenia and bone lesions or impending fractures. Magnetic resonance imaging (MRI) is more sensitive than the bone survey, can identify early bone disease and extramedullary disease, and can distinguish myeloma from normal marrow, helping in quantifying the extent of the disease. Positron emission tomography coupled with computed tomography (PET-CT) detects metabolically active intramedullary or extramedullary myeloma foci and lytic bone lesions. The most common staging system for myeloma is the International Staging System, based on two parameters, serum β2-microglobulin (β2M) and albumin; three stages are defined and correlate with patient outcome. Serum levels of β2M, C-reactive protein, number of circulating plasma cells and their labeling-index are related to patient prognosis. Other prognostic factors include the presence of specific chromosomal abnormalities (deletion of chromosome 17p and loss of chromosome 1p/gain of chromosome 1q), altered gene expression profiling, as well as MRI and PET-CT abnormalities. The introduction of the immunomodulatory drugs (IMiDs) thalidomide and lenalidomide and the proteasome inhibitor bortezomib have increased the 5-year relative survival rate to 45 percent, with some patients achieving long-term survival of more than 10 years. Melphalan-based autologous hematopoietic stem cell transplantation in combination with novel agents can achieve remarkable results in patients with good risk myeloma; patients who are not eligible for autologous transplant have traditionally been treated with melphalan and prednisone; novel agents such as bortezomib and lenalidomide are also active and well-tolerated also in this population. Consolidation and maintenance regimens based on lenalidomide as single agent or in combination with bortezomib have been evaluated to extend the duration of complete remission following autologous stem cell transplant. Finally, several novel agents can be used in the setting of relapsed/refractory disease including new proteasome inhibitors (carfilzomib, ixazomib, and marizomib), IMiDs (pomalidomide), histone deacetylase inhibitors (vorinostat, panabinostat, and ricolinostat), and monoclonal antibodies (daratumumab and elotuzumab).
Acronyms and Abbreviations
auto-HSCT, autologous hematopoietic stem cell transplantation; BMSCs, bone marrow stromal cells; β2M, β2-microglobulin; CALGB, Cancer and Leukemia Group B; CR, complete response; CRD, carfilzomib, lenalidomide, and dexamethasone; CT, computed tomography; CyBorD, cyclophosphamide, bortezomib, and dexamethasone; del, deletion; DLI, donor lymphocyte infusion; EBMT, European Bone Marrow Transplant Group; ECM, extracellular matrix; ECOG, Eastern Cooperative Oncology Group; EFS, event-free survival; EMP, extramedullary plasmacytoma; FISH, fluorescence in situ hybridization; GVHD, graft-versus-host disease; GVM, graft-versus-myeloma effect; IGF-1, insulin-like growth factor; ISS, International Staging System; LCDD, light-chain deposition disease; MG, essential monoclonal gammopathy; MGUS, monoclonal gammopathy of undetermined significance; MIP, macrophage inflammatory protein; MP, melphalan-prednisone; MRD, minimal residual disease; MRI, magnetic resonance imaging; MTD, maximum tolerated dose; ONJ, osteonecrosis of the jaw; ORR, overall response rate; OS, overall survival; PET, positron emission tomography; RVD, lenalidomide, bortezomib, and dexamethasone; SCID, severe combined immunodeficiency; sCR, stringent complete response; SMM, smoldering multiple myeloma; SOP, solitary osseous plasmacytoma; TGF-β, transforming growth factor-β; TTP, thrombotic thrombocytopenic purpura; VGPR, very good partial response; VTE, venous thromboembolism.
DEFINITION AND HISTORY
Myeloma is a disease characterized by clonal expansion of malignant plasma cells that accumulate in the marrow, leading to anemia and associated cytopenias, hypogammaglobulinemia, osteolytic bone disease, hypercalcemia, and renal dysfunction.1 Symptoms are a result of tumor mass effects, cytokine release by myeloma cells, marrow stroma or bone cells, or deposition of myeloma proteins into target organs (amyloid light-chain [AL] amyloidosis and light-chain deposition disease [LCDD]). Myeloma is part of a spectrum of diseases called plasma cell dyscrasias that include the following conditions: essential monoclonal gammopathy (MG; also known as monoclonal gammopathy of undetermined significance [MGUS]), smoldering myeloma (Chap. 106); solitary plasmacytoma, located in the bone as isolated lytic lesion or in the soft tissues; macroglobulinemia (Chap. 109); AL amyloidosis and LCDD (Chap. 108); and very rare disorders, such as osteosclerotic myeloma (POEMS [polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes] syndrome), Castleman disease, and heavy chain diseases (α-heavy-chain disease, μ-heavy-chain disease, and γ-heavy chain disease) (Chap. 110). Myeloma derives from cells with plasma cell morphologic features, capable of producing immunoglobulin molecules (Chap. 75), which can be detected in the serum and/or urine by electrophoresis and immunofixation. By definition, plasma cell dyscrasias result from the expansion of monoclonal cells, with resultant monoclonal protein secretion; however, oligoclonal and polyclonal protein abnormalities accompany some conditions, such as Castleman disease.
EPIDEMIOLOGY
Myeloma represents the second most common hematologic cancer, accounting for 1.4 percent of all cancers and 10 percent of hematologic malignancies, with a prevalence of 83,367 people in 2011.2 The American Cancer Society has estimated that 24,050 new myeloma cases were diagnosed in the United States in 2014, of which approximately 11,090 will die. Most of the patients are diagnosed among people ages 65 to 74 years, with a median age at onset of 69 years; only 4 percent of cases occur before age 45 years. Men are affected more frequently than women (1.6:1 ratio) and individuals of African descent have twice the prevalence of myeloma as those of European descent. Conversely, individuals of Japanese and Spanish (Latino) descent have very low prevalence rates.3,4,5 Myeloma is always preceded by a condition called MG, as has been demonstrated by long-term followup studies of a cohort of more than 70,000 banked serum samples from healthy subjects and from an independent military cohort.6,7 MGUS is present in 3.2 to 4.0 percent of the general population,8 developing years before the diagnosis of myeloma, at a rate of approximately 1 percent per year (Chap. 106).9
The etiology and the mechanisms of myeloma progression are still largely unknown.1 Several epidemiologic reports have demonstrated an increased risk of myeloma or MG (up to fourfold) in first-degree relatives of individuals affected by plasma cell dyscrasias.10 Moreover, myeloma is associated with an increased risk of prostate cancer, melanoma, non-Hodgkin lymphoma, and chronic lymphocytic leukemia.11 More than 100 myeloma families have been described from different geographic areas12,13,14; in one family, a germline mutation of the CDKN2A (p16) gene together with loss of heterozygosity of the other allele was identified as a rare low-penetrance genetic risk.13 Genome-wide association studies have identified six single nucleotide polymorphisms (SNPs) at chromosomes 2p23.3, 3p22.1, 3q26.2, 6p21.33, 7p15.3, 17p11.2, and 22q13.1 that are associated with risk of myeloma. The identified genes (DNMT3A, ULK4, TERC, PSORS1C1, CDCA7L/DNAH1, TNFRSF13B, and CBX7) have never been validated as myeloma-driver genes.15,16 These same SNPs have also been independently associated with development of MG, with the risk increasing with the number of alleles carried.17 Moreover, the presence of hyperphosphorylated paratarg-7 (pP-7) carrier status has been reported as an autosomal dominantly inherited risk factor for MG and myeloma in a European population and in MG/myeloma cases in African Americans.18,19
Some epidemiologic studies, including a cohort study of Swedish and Finnish twins and a meta-analysis,20,21 have shown an association between high body mass index and risk of myeloma.22,23 Specifically, obese individuals have higher levels of cytokines, such as interleukin (IL)-6 and insulin-like growth factor (IGF-1), which also are produced by adipocytes and are potent growth factor for myeloma cells.24 No consistent associations have been observed with any particular diet, alcohol consumption, or smoking.25 Occupational exposure to pesticides, organic solvents (benzene, petroleum derivatives, styrene) or chronic radiation have been alleged to be associated with myeloma in some studies,25,26 but refuted by others. Furthermore, the use of thorium dioxide (thorotrast), a contrast medium used in the 1950s for angiography, increases the risk of plasmacytomas up to fourfold.27 Exposure to acute radiation, as in atomic bomb survivors increases the overall rate of MG or myeloma after 15 to 20 years,28 and accelerates MG transformation to myeloma.29 People exposed to fresh wood, wood dust, or working in saw mill factories had an increased risk of myeloma in some studies.30,31 Finally, associations between myeloma risk and autoimmune diseases (especially rheumatoid arthritis32,33 or pernicious anemia34) or infections (HIV and hepatitis C35,36) have been proposed, based on a retrospective study among American male military veterans,37,38 suggesting an immune-mediated mechanism for malignant transformation. Human herpes virus 8 DNA sequences responsible for Kaposi sarcoma and Castleman disease pathogenesis39 have been reported by some investigators in marrow dendritic cells of myeloma patients, although other studies suggest that this is an epiphenomenon.40,41
ETIOLOGY AND PATHOGENESIS
Myeloma cells derive from postgerminal–center marrow plasmablasts/plasma cells (Fig. 107–1). Myeloma cell immunoglobulin heavy chain (IGH) variable genes present somatic mutations in the absence of intraclonal variation or ongoing somatic hypermutation, indicating antigen-contact selection in the germinal center.42,43 The existence of a myeloma stem cell, that is a precursor with self-renewal capacity, has been proposed for a long time, given myeloma’s low proliferative index and clonogenic efficiency. However, this remains a matter of debate.44 Myeloma is a multistep process, always preceded by a MG phase.6,7 MG cells share several similarities with myeloma, including a similar prevalence of hyperdiploidy and of the three primary IGH rearrangements [t(6;14), t(11;14), and t(14;16)]. However, chromosome 13 deletions, RAS mutations and non–immunoglobulin (Ig)-locus associated MYC translocations are more frequent in myeloma.43 Indeed, the development of myeloma seems to necessitate an immortalizing event, such as a primary IGH translocation, an oncogene activation, or deregulation of a tumor suppressor, to occur in the germinal center during the switch recombination or somatic hypermutation, resulting in uncontrolled expansion of a long-lived plasmablast/plasma cell.42 In early stages, myeloma cells are dependent on the growth support provided by bone marrow stromal cells (BMSCs) (intramedullary phase), but can become independent of their medullary environment at late stages (plasma cell leukemia). However, 15 to 70 percent of newly diagnosed myeloma patients, using conventional morphology techniques45,46,47,48,49,50,51 or multiparametric flow cytometry52 have circulating clonotypic myeloma cells (circulating tumor cells [CTCs]) in their blood, suggesting the presence of a “metastatic”/dissemination process that disseminates the disease hematogenously.53 Moreover, the presence of CTCs in MG is a risk factor for myeloma progression,54,55 as well as a poor prognostic factor in newly diagnosed or relapsed/refractory myeloma patients.56,57 Myeloma CTCs share a similar phenotype to marrow myeloma cells, but are more quiescent, have better in vitro clonogenic capacity and have lower expression of integrin and adhesion molecules (including CD138) making them less dependent on marrow niches.58
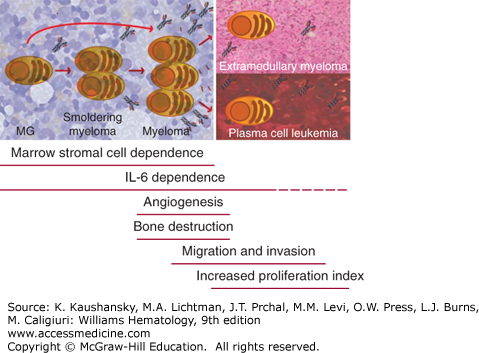
Figure 107–1.
Myeloma stages, from essential monoclonal gammopathy (MG) to plasma cell leukemia. Myeloma evolves from a benign condition called essential monoclonal gammopathy (or monoclonal gammopathy of undetermined significance), with an annual rate of progression of 1 percent. In some patients, a stage called smoldering myeloma is sometimes evident, where there is a higher number of monoclonal plasma cells in the marrow, but still absence of symptoms. At early stages during the so-called intramedullary phase, myeloma cells are totally dependent on marrow microenvironment to survive and on interleukin (IL)-6 and other cytokines. During progression, myeloma cells can acquire the capability of growing without microenvironmental support and localize to other tissues (extramedullary disease) or circulate in the blood (secondary plasma cell leukemia). Active myeloma is characterized by onset of angiogenesis and bone lytic lesions in contrast to MG or smoldering myeloma; during late stages there is an increase in migration and invasion capabilities, as well as high proliferative rates.
Myeloma is a heterogeneous disease with a complex genetic landscape, characterized by several numerical and structural aberrations, including abnormal karyotypes, chromosomal translocations and copy-number changes (Fig. 107–2 and Table 107–1).
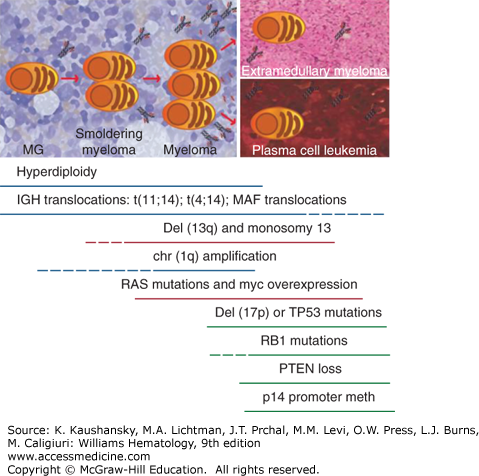
Figure 107–2.
Genomic aberrations, including karyotype abnormalities, chromosomal translocations, and copy number variations in essential monoclonal gammopathy (MG), myeloma, and plasma cell leukemia. Myeloma cells are characterized by several genomic aberrations, which combine differently in distinct patients. Hyperdiploidy and immunoglobin heavy-chain (IGH) translocations [t(11;14), t(4;14) and MAF translocations] are already present in the MG phase, a benign condition that can evolve to active myeloma with a rate of 1 percent per year. These abnormalities are not considered driver events in myeloma. Conversely, several groups have proposed other aberrancies, such as MYC translocations and increased MYC mRNA levels or RAS mutations as transforming events, because they are rare in MG and smoldering myeloma but common in myeloma. Also chromosome gains and losses, including deletion of chromosome 13q or monosomy 13, deletion of chromosome 1p, and amplification of chromosome 1q21 are seen more frequently in active myeloma, even though their role in myeloma progression is still not totally elucidated. Deletion of chromosome 17p or TP53 mutations are rare at diagnosis, but present in advanced/relapsed settings, being associated with reduced response to treatment and unfavorable patient outcomes. The acquisition of independence from support by the marrow microenvironment is a feature of advanced myeloma, possibly leading to plasma cell accumulation in various organs (extramedullary disease) or in the blood (plasma cell leukemia). PTEN losses, methylations of p14 promoter, and RB1 inactivations are reported more frequently in plasma cell leukemia, suggesting a role in the development of extramedullary growth.
| Genetic Lesion | MG | Myeloma | Plasma Cell Leukemia |
|---|---|---|---|
| Hyperdiploidy | 50% | 60% | 20% |
| t(11;14) | 5–10% | 20% | 25–60% |
| t(4;14) | 2–3% | 15% | 15–25% |
| MAF translocations | 5% | 15–35% | |
| Del(13q)/Monosomy 13 | 20% | 50–60% | 60–80% |
| Del(1p) | 4% | 7–40% | |
| Chr 1q21 amplification | 40% | 70% | |
| Cyclin D dysregulation | 60% | 80% | |
| RAS mutations | <5% | 30–50% | 30% |
| FAM46C, DIS3 | 10–21% | ||
| NF-κB activating mutations and CNVs | 15–20% | ||
| IGH MYC rearrangements | 1–2% | 15% | 30–50% |
| UTX deletions and mutations | 30% | ||
| TP53 inactivations (mutations + del(17p)) | 5% | 10–20% | 20–80% |
| p18 and/or Rb inactivation | <5% | 25–30% | |
| p14 promoter methylation | <5% | 25–30% | |
| PTEN loss | 0% | <2% | 8–33% |
Traditionally, myeloma patients have been divided into two subgroups: hyperdiploid cases with more than 46 but less than 76 chromosomes (34 to 60 percent of myeloma); and nonhyperdiploid cases, which include individuals with a hypodiploid (up to 44 to 45 chromosomes), pseudodiploid (44/45 or 46/47 chromosomes with gains or losses), and near-tetraploid karyotype.59,60,61 Hyperdiploid patients, normally IgG kappa-type with bone involvement, show gains of odd-numbered chromosomes, including trisomies of chromosomes 15, 9, 5, 19, 3, 11, 7, and 21 (ordered by decreasing frequency), and have a favorable prognosis that can however be affected by the concomitant presence of additional abnormalities such as chr11 or chr1q gains or chr13 loss.62,63 Fluorescence in situ hybridization (FISH) analysis is employed to detect five major primary IGH translocations in myeloma,64 which occur more frequently in nonhyperdiploid patients (85 percent vs. <30 percent).65 Primary translocations are caused by errors during normal DNA recombination in isotype class switching of terminally differentiated B cells. Conversely, IGH translocations involving chromosome 8p24 and 11q13 (called secondary translocations) result from errors in somatic hypermutation processes.42 All of the translocations induce increased constitutive expression of specific oncogenes by their juxtaposition to immunoglobulin enhancer elements. The most frequent translocation (20 percent of cases) is t(11;14)(q13;q32),66,67,68 leading to upregulation of cyclin D1, a crucial promoter of G1-to-S transition via cyclin-dependent kinase (CDK)-4 or CDK6.69,70 Rarely, cyclin D2 and cyclin D3 can be rearranged via t(12;14) (<1 percent) or t(6;14) translocations (2 percent of myeloma patients), respectively.71 Even in the absence of translocations, cyclins D1, D2, and D3 are often upregulated, creating specific patient subgroups with different prognoses.72 Specifically, the CD-1 subgroup (cyclin D1-high) responds well to treatment and has an increased frequency of early relapse but also has an excellent long-term survival, while the CD-2 subgroup (cyclin D3-high) exhibits a lymphoplasmacytoid morphology. Translocation t(4;14), a poor prognostic factor, pairs MMSET/WHSC1, a nuclear SET DOMAIN protein with FGFR3 (fibroblast growth factor receptor), an oncogenic tyrosine kinase receptor in 15 percent of patients, often in association with chromosome 13 abnormalities.73,74,75,76 MMSET is an H3K4-, H3K27-, H3K36-, and H4K20-specific histone methyltransferase, that causes global changes in chromatin status, favoring myeloma cellular and clonogenic growth, adhesion, and tumorigenicity,75 while FGFR3 promotes myeloma cell proliferation via RAS-MAPK (mitogen-activated protein kinase) and STAT (signal transducer and activator of transcription) pathways.77 Additionally, activating FGFR3 mutations, mutually exclusive with RAS mutations, have also been reported in a small fraction of patients.78,79 MAF translocations, which include t(14;16) overexpressing c-MAF, t(14;20) which deregulates MAFB, and t(8;16) involving MAFA are relatively rare (5 percent, 2 percent, <1 percent of cases, respectively) but associated with poor prognosis.80 c-MAF, MAFA, and MAFB are all transcription factors involved in proliferation, responsiveness to IL-6 and BMSC-MM (multiple myeloma) adhesion,81 promoting cell-adhesion-mediated drug resistance (CAM-DR), via integrin α7/E-cadherin interactions.82 The prevalences of these three primary IGH rearrangements [t(6;14), t(11;14), and t(14;16)] are similar in MG, indicating the need of additional transforming events to precipitate active myeloma.83,84,85
Array comparative genomic hybridization (aCGH) analysis demonstrates numerous copy number alterations (CNAs) in myeloma cells. Deletion of chromosome 13, deletion of chromosome 17p13, and amplification of chromosome 1q21 are genomic aberrations associated with poor prognosis in myeloma patients.86,87 Deletion of chromosome 13 affects 50 to 60 percent of newly diagnosed myeloma, is more frequent in the nonhyperdiploid group (>70 percent) in comparison to the hyperdiploid group and often cooccurs with t(4;14) or t(14;16) translocations.88 Among other genes, RB1 and the miRNA-15a/16–1 cluster are deregulated in this context and may play a role in myeloma pathogenesis. Despite being traditionally associated with a poor prognosis, the adverse impact of isolated chromosome 13 deletion is now controversial,89 in view of the good response of such patients to bortezomib-based regimens and the close association of del13 with the t(4;14)(p16;q32) and hypodiploid karyotype.
Deletions of chromosome 17p involving the TP53 locus are rare in newly diagnosed myeloma (5 to 10 percent), more common in relapsed and refractory cases (20 to 40 percent), and inevitably associated with negative prognosis, causing early relapse in patients treated with or without autologous stem cell transplantation. Mutations in TP53 are also often present on the second allele.90,91 Despite having an unfavorable prognosis, regimens containing bortezomib can increase the median progression-free survival (PFS) and 3 years of patients with TP53 mutations (17 percent to 69 percent [P = 0.028]) in comparison to non–bortezomib-containing regimens, as shown by the HOVON-65/GMMG-HD4 trial.92 1q21 amplification is detected by FISH in approximately 40 percent of newly diagnosed myeloma and in 70 percent of relapsed myeloma, and negatively affects overall survival (OS), with a cumulative effect based on the number of 1q21 locus copies.93 Possible target genes of this lesion are CKS1B, a protein that regulates cyclin-dependent protein kinases, PSMD4, a proteasome subunit modulating response to bortezomib treatment, MCL1 or BCL9.93,94,95 Interestingly, a reported jumping translocation of 1q12 (JT1q12) can have a receptor chromosome TP53 genomic locus, causing simultaneous gain of 1q21 and deletion of 17p.96 Additionally, deletions of 1p, present in 7 to 40 percent of patients, are linked to reduced PFS and OS despite autologous stem cell transplantation.97,98,99 TP73, LAPTM5, CDKN2C, a CDK inhibitor which interacts with CDK4 or CDK6 to regulate G1/S phase, MTF2, TMED5, and FAM46C are candidate genes of the deletion. FAM46C loss or FAM46C mutations (evident in 15 percent of patients) are especially associated with shortened survival (median OS 25.7 months vs. 51.3 months, P = 0.004).97,100 The biologic function of FAM46C is uncertain, although some data suggest it is related to mRNA stabilization. Other mutations include MYC rearrangements involving unbalanced translocations and insertions, small duplications, amplifications, and inversions on chromosome 8p24101,102,103,104; homozygous deletions of 11q22 locus resulting in loss of YAP1, BIRC3, and BIRC2 genomic region105,106,107; chromosomes 4, 14, and 16 aberrations that disrupt FGFR3, WWOX, and CYLD; deletions or amplifications of chromosome 6; and homozygous deletions of Xp11.2 locus,62,63 involving UTX, an histone H3 lysine 27 (H3K27) demethylase, which is also mutated in 10 percent myeloma,100 are also common. The “purpose” of these multiple plasmin inhibitors is to guard against premature plasmin activation and subsequent degradation of fibrinogen, until intravascular fibrin begins to appear.
Somatic Mutations and Interclonal Diversity Myeloma evolves in a stepwise process, transforming from MG to smoldering myeloma to overt myeloma, where mutations accumulate, conferring either a growth advantage (driver mutations) or are functionally irrelevant (passenger mutations). So far, more than 300 myeloma patient DNA samples have been sequenced using whole-genome sequencing or whole-exome sequencing approaches.100,108,109,110,111,112,113 Specifically, 11 genes are commonly mutated in myeloma reaching a standard significant threshold.112 Five of them (KRAS, NRAS, FAM46C, DIS3, and TP53) are relatively frequent.100,108 Approximately 30 to 50 percent of newly diagnosed patients have RAS mutations at codons 12 or 13,82,114,115,116,117 often in association with t(11;14), but mutually exclusive with t(4;14) that constitutively activates the MAPK pathway via FGFR3 upregulation.115,118,119 Because RAS mutations are rare in MG, they are considered a possible driver to progression.120 Conversely, TP53 mutations are a late event in myeloma,91,121,122 an independent poor prognostic factor,123 and are associated in one-third of patients with concomitant hemizygous deletion of chromosome 17.90,124 Mutations in FAM46C and DIS3, genes possibly involved in RNA processing, are present in 10 to 21 percent of patients, often coupled with loss-of-heterozygosity in the remaining allele. Other significant genes are BRAF (4 percent of patients), TRAF3, CYLD, RB1, PRDM1, and ACTG1. TRAF3 and CYLD mutations, together with homozygous deletions in BIRC2/BIRC3, NIK overexpression and mutations in other genes (CARD11 and MYD88) contribute to constitutively activation of the NF-κB pathway.100,105,107,112 Genes involved in protein homeostasis, the unfolded protein response, or lymphoid/plasma cell development, such as PRDM1 involved in plasmacytic differentiation, and XBP1, IRF4, LRRK2, SP140, and LTB form a cluster of genes mutated in myeloma. Other recurrent mutated genes are ROBO1, a transmembrane receptor involved in β-catenin and MET signaling; EGR1 transcription factor; FAT3, a transmembrane protein belonging to cadherin superfamily; and histone-modifying genes (MLL, MLL2, MLL3, WHSC1/MMSET, WHSC1L1, and UTX, among others).100,108,112 Plasma cell leukemia patients possess different aberrancies, including p14ARF promoter methylation, PTEN loss, RB1 mutations and higher rates of TP53 mutations and deletions.125 Finally, a novel intriguing concept is the idea of intratumor heterogeneity in myeloma, where different subclones can emerge and become predominant following different mechanisms of evolution, including linear, branching, parallel or convergent evolution.108,126 Clonal diversity is indeed a fundamental process akin to Darwinian selection, favoring cancer progression and adaptation to therapy. Next-generation sequencing analyses show that most patients have a subclonal structure at diagnosis, with one predominant clone and several others which can reappear at different stages or following treatment.108,110,111 Gene-expression profiling of myeloma cells helps categorize patients into distinct subgroups127,128 and can predict therapeutic responsiveness to specific drugs, although the role of specific genetic signatures in risk stratification is still under debate.129
The marrow microenvironment is composed of extracellular matrix (ECM) proteins, such as fibronectin, collagen, laminin and osteopontin; and cells, including hematopoietic stem cells, BMSCs, and endothelial cells, as well as osteoclasts and osteoblasts (Fig. 107–3) (Chap. 5).130,131,132,133 Myeloma cells physically interact with ECM proteins and accessory cells to gain growth, survival, and drug resistance advantages. Among others, CD44, very-late antigen 4 (VLA4), neuronal adhesion molecule (NCAM), intercellular adhesion molecule (ICAM)-1, and syndecan 1 (CD138) mediate the adhesion of myeloma cells to the marrow and ECM, activating signaling pathways, such as nuclear factor-κB (NF-κB), to obtain CAM-DR to conventional chemotherapy.134,135 In particular, CD138 (syndecan-1) is a transmembrane heparan sulphate bearing proteoglycan, expressed during the plasma cell stage of B-cell maturation, that can bind to type I collagen inducing expression of metalloproteinases, and promoting bone resorption and invasion.136 Moreover, CD138 can be shed in the ECM, trapping growth-promoting and proangiogenic cytokines.137,138 Increased soluble CD138 levels correlate with tumor burden and poor outcomes.139 The SDF-1/CXCR4 axis regulates specific homing of myeloma cells to the marrow, but also mobilization or marrow egress, being possibly accountable for the multifocal marrow localization and blood circulation of myeloma cells.140,141 Moreover, other chemokine receptors, such as CXCR3, CCR1, CCR2, and CCR5 can be expressed by myeloma conferring different migration capabilities to medullary and extramedullary cells.142 Accessory cells (BMSCs, endothelial cells, osteoclasts, and osteoblasts) secrete factors including IL-6,143,144,145,146 IGF-1,147,148,149 vascular endothelial growth factor (VEGF),150,151 tumor necrosis factor-α (TNF-α),152 fibroblast growth factor (FGF),153 stromal cell-derived factor 1α (SDF-1α),141 and B-cell activating factor (BAFF)154; all capable of promoting expression of survival factors such as NF-κB.155 IL-6 and other survival signals also induce phosphatidylinositol 3′-kinase (PI3K)/AKT,156,157 STAT3 and MAPK signaling (Fig. 107–3).133 BMSCs, myeloma cells and osteoclasts also secrete growth factors and cytokines such as VEGF, basic fibroblast growth factor (bFGF), and IL-8, to promote marrow angiogenesis, which increases delivery of oxygen and nutrients to myeloma cells. Moreover, the same endothelial cells produce growth factors (IL-6 and IGF-1), to favor plasma cell survival150,151,158,159 and express deregulated genes important for ECM and bone remodeling, cell adhesion, migration and resistance to apoptosis.160 The degree of marrow angiogenesis, as assessed by microvessel density (MVD) is higher in active myeloma compared with MG161 and is also related to myeloma proliferation and infiltration, negatively affecting patient prognosis.162 Also lymphoid and myeloid cells are part of marrow microenvironment and can modulate myeloma survival. Myeloid cells, such as macrophages, mast cells and neutrophils control both pro- and antiinflammatory responses and regulate antigen presentation.163 For instance, a specific group of myeloid cells, named myeloid-derived suppressor cells (MDSCs),164 are highly represented in the marrow of myeloma individuals in comparison to healthy persons, and increase with disease progression, facilitating tumor development, growth, and immune escape, by blocking T-cell (CD8+ T and natural killer [NK] T) antitumor immune responses.165
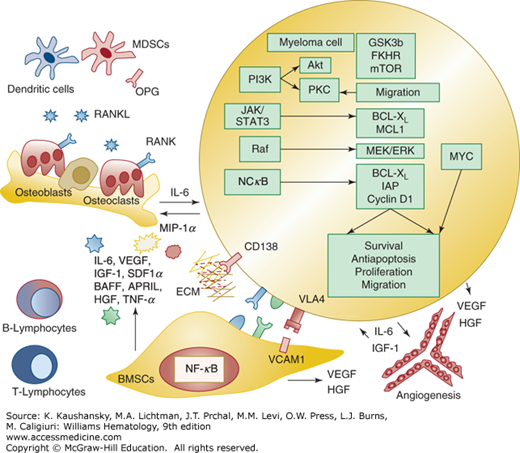
Figure 107–3.
Myeloma cell interaction with extracellular matrix (ECM) and accessory cells in the marrow. Myeloma cells require support from bone marrow stromal cells (BMSCs) during early stage disease. Adhesion between myeloma cells and BMSCs favors myeloma cell survival, growth, and migration via release of cytokines (IL-6, VEGF, IGF-1, SDF1α, BAFF, APRIL, HGF, TNF-α) from both myeloma cells and BMSCs. Among others, extracellular signal-regulated kinase (ERK); Janus kinase 2 (JAK2)–signal transducer and activator of transcription 3 (STAT3); phosphatidylinositol 3′-kinase (PI3K)–Akt; nuclear factor-κB (NF-κB) and MYC are constitutively active in myeloma, promoting transcription or activation of important targets, including cytokines (IL-6, IGF-1, VEGF), antiapoptotic proteins (BCL-XL, IAP, MCL1), cell-cycle modulators (cyclin D1) and proteins involved in migration, invasion, and autophagy. NF-κB activation in both myeloma and BMSCs upregulates adhesion molecules (VCAM1, VLA4) to promote reciprocal binding. Proangiogenic factors, including VEGF and HGF are released from myeloma cells, BMSCs and marrow endothelial cells, to promote neoangiogenesis and increase delivery of oxygen and nutrients to tumor cells. Cells from the innate and adaptive immune response, including B lymphocytes, T lymphocytes, dendritic cells, and myeloid-derived suppressor cells, are also modulated by myeloma cells, creating an immunosuppressive microenvironment that promotes tumor survival and reduces antigen presenting capabilities. Receptor activator of NF-κB ligand (RANKL) and MIP-1α are produced by BMSCs and myeloma cells and trigger osteoclast activation via RANK receptor. Osteoprotegerin (OPG), a decoy receptor for RANKL secreted by osteoblasts and BMSCs to block RANKL–RANK ligand interaction and inhibit osteoclastogenesis, is reduced in myeloma patients. APRIL, a proliferation-inducing ligand; BAFF, B-cell activating factor; HGF, human growth factor; IGF-1, insulin-like growth factor; IL, interleukin; MIP, macrophage inflammatory protein; SDF1α, stromal cell derived factor 1α; TNF, tumor necrosis factor; VCAM, vascular cell adhesion molecule; VEGF, vascular endothelial growth factor; VLA, very-late antigen.
The presence of osteolytic bone lesions, bone pain, increased risk of pathologic fractures and generalized bone loss (or osteoporosis) is a well-defined feature of myeloma.166 Indeed, as myeloma burden increases, an imbalance between osteoblast and osteoclast activities ensues, with suppression of bone formation by osteoblasts and uncoupled activation of osteoclasts (Fig. 107–4).167,168,169 The ligand for receptor activator of NF-κB (RANKL) binds to RANK receptor to stimulate osteoclast differentiation, formation and survival170; myeloma cells produce RANKL and upregulate RANKL expression in BMSCs and osteoblasts via direct contact interaction, signaling induction171,172,173 or production of IL-7. Moreover, they promote suppression of osteoprotegerin (OPG),174,175,176 a decoy receptor that normally prevents RANK–RANKL interaction177,178 via soluble factors, integrin α4β1-vascular cell adhesion molecule (VCAM)-1 interaction,179 production of Dickkopf-1(DKK1),180 or inactivation by syndecan-mediated internalization into myeloma cells.181 Interestingly, OPG levels are decreased in the serum of myeloma patients and correlate with lytic bone lesion development182; a high RANKL-to-OPG ratio is associated with worse prognosis.183 Recombinant OPG constructs, soluble RANK, OPG peptidomimetics175,178,184,185 and an anti-RANKL antibody, denosumab,186,187,188,189 have been developed to modulate the RANKL/OPG axis and reduce osteoclast activity in myeloma. Macrophage inflammatory protein (MIP)-1α, or chemokine C-C motif ligand, is also produced by myeloma cells and promotes maturation of precursor cells into osteoclasts190,191,192; MIP-1α signals via CCR1 and CCR5 on osteoclasts and can further upregulate RANKL in stromal cells.190 MIP-1α levels are elevated in myeloma patients,193,194 whereas MIP-1α silencing or blockade of CCR1 reduces bone disease in in vitro or animal models. IL-6,195 parathyroid hormone-related peptide (PTHrP),196,197 annexin II,198 and the ephrinB2/EphB4 axis199 also promote bone reabsorption. Osteoblast suppression is another major player in myeloma bone disease: WNT signaling antagonists, including DKK1, frizzled related protein-2 (FRP-2),200 and sclerostin (SOST),201,202 interfere with osteoblast maturation. DKK1 is expressed by myeloma cells and can upregulate RANKL levels in osteoblasts, increasing osteoclast activity.203,204,205 DKK1 levels are increased in the serum of myeloma patients,206 and anti-DKK1 antibodies have been tested in animal studies207,208,209,210 and are currently employed in clinical trials. Finally, high levels of Activin A, a member of transforming growth factor-beta (TGF-β) superfamily, IL-3 and IL-7 (via RUNX2/CBFA1 blockade) can inhibit bone formation and promote bone reabsorption.211,212,213 Furthermore, the bone niche itself supports myeloma cell survival and prevents TNF-α–mediated apoptosis by producing various molecules.167 Indeed, bisphosphonates not only block osteoclasts and modulate osteoblasts, but have an effect on tumor burden.214 A similar effect is reported with OPG peptidomimetics and RANKL constructs in in vivo xenograft models.215 Hence, bisphosphonates, especially zoledronic acid, are currently used in the clinic to reduce bone disease,216,217 but are also associated with an increase in OS when compared to placebo based on a meta-analysis.218 Markers of bone resorption and formation correlate with the extent of osteolytic disease.219 Specifically, urine levels of pyridinoline (PYD) and deoxypyridinoline (DPD) crosslinks and serum levels of tartrate-resistant acid phosphatase isoform 5b (TRACP-5b), a resorption marker only produced by activated osteoclasts and of collagen degradation products, including the N-terminal crosslinking telopeptide of type I collagen (NTX), are elevated in myeloma patients compared with healthy controls and can predict early progression of bone disease in myeloma. Conversely, bone formation markers, such as bone alkaline phosphatase (bALP) and osteocalcin (OC), are reduced.219
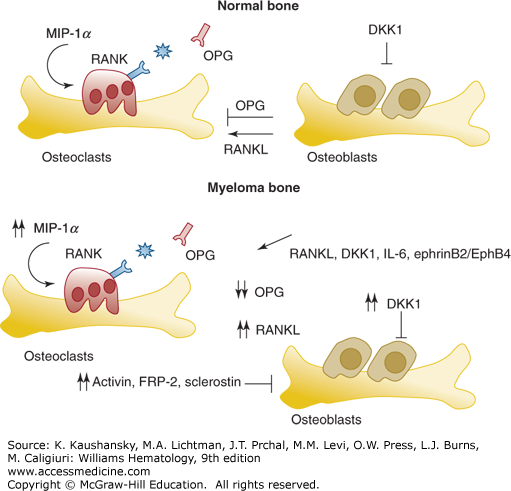
Figure 107–4.
Mechanism of bone remodeling in normal conditions and in the presence of myeloma cells. The major factors affecting osteoclast and osteoblast activation, and thus the balance between bone formation and bone reabsorption, are illustrated in the upper panel. Receptor activator of nuclear factor-κB (RANK) receptor/receptor activator of nuclear factor-κB ligand (RANKL) and macrophage inflammatory protein (MIP)-1α stimulate osteoclastogenesis and osteoclast activity, while osteoprotegerin (OPG) acts a decoy receptor for RANKL, reducing its action. DKK1 (Dickkopf-1) is an inhibitor of osteoblast activity. In the presence of myeloma cells in the bone, the normal balance between osteoblasts and osteoclasts is totally inverted. Specifically, myeloma cells secrete factors to promote osteoclast activation, a result of upregulation of RANKL and MIP-1α, and to inhibit osteoblasts. Increased levels of DKK1, activin, FRP-2 (frizzled related protein-2), and sclerostin are evident in myeloma patients. In red are marked cytokines or receptors used as targets to treat myeloma bone disease. IL, interleukin.
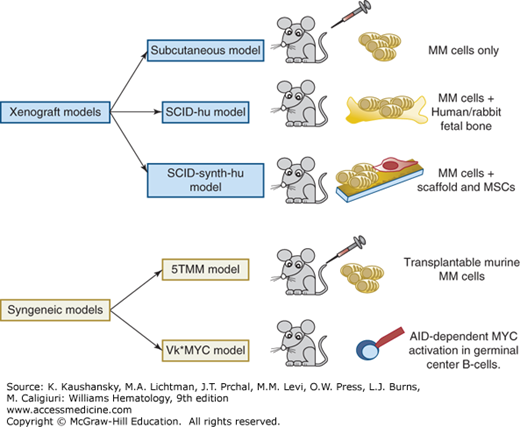
Novel therapies need to be tested in preclinical in vitro/vivo models capable of mimicking the role of human marrow microenvironment. In in vitro settings, myeloma cells are cocultured in liquid or semisolid systems together with different cytokines (IL-6, IGF-1, TNF-α) or with autologous BMSCs from patients. However, these systems do not truly recapitulate the marrow microenvironment220,221,222 and in vivo models are necessary. Two main types of myeloma animal models have been exploited to study human myeloma biology and response to treatment: xenogeneic models in immunodeficient and humanized mice or syngeneic tumor models (Fig. 107–5).223 The xenogeneic models require subcutaneous injection of tumor myeloma cells in severe combined immune deficiency (SCID) or nonobese diabetic (NOD)/SCID mice, which are immunocompromised. These models lack the complex marrow–myeloma interaction, but can still be used to explore myeloma homing and novel drugs. Conversely, the SCID-hu or the SCID-rab mice models recreate a look-alike microenvironment, able to sustain myeloma cell growth.224 Specifically, primary myeloma cells or myeloma cell lines are grown in human fetal bone (the SCID-hu model) or rabbit bone (SCID-rab model), later implanted in SCID mice. Myeloma cells grow inside the implanted bones, or disseminate to the outer surface of the implanted bone, if they derive from patients with extramedullary disease. Moreover, these mice have circulating monoclonal immunoglobulins, osteolytic lesions, suppression of bone formation near myeloma foci and angiogenesis.224,225 Studies propose novel xenogeneic models, where three-dimensional bone-like scaffolds are used instead of human fetal bone or rabbit bone.226,227 Specifically, these scaffolds are internally coated with murine/human BMSCs or human mesenchymal stromal cells and then implanted in SCID or RAG2–/–γc–/– mice, to recapitulate the autologous marrow microenvironment in vivo. Myeloma cells are then loaded directly inside the scaffold or injected intracardiac to mimic myeloma homing. These models are more suitable for studying the microenvironment and also for drug testing, affording the opportunity to evaluate different stages or type of disease. Syngeneic tumor models include transplantable murine models like 5T33,228,229 which are mostly representative of aggressive late-stage disease, as well as genetically engineered mouse models, such as the Vk*MYC mouse,230 generated from the C57BL/6 mouse strain, which has a high incidence of spontaneous monoclonal gammopathy with aging, after activation-induced cytidine deaminase–dependent MYC activation in germinal center B-cells. This model exhibits high penetrance, clonal malignant plasma cells, which produce isotype class-switched immunoglobulins, a similar histology and immunophenotype to human myeloma, and delayed onset of renal failure, bone lesions, and anemia. The Vk*MYC mouse model has been used for drug testing, strongly paralleling the drug activity observed in myeloma patients.230
Figure 107–5.
Preclinical models of myeloma. Xenogeneic and syngeneic models have been developed to study myeloma biology and test novel therapies. In xenogeneic models, human myeloma (MM) cells are injected subcutaneously, inside human/rabbit fetal bone or into synthetic scaffolds, previously coated with mesenchymal stromal cells (MSCs). In the last two types of models, a look-alike microenvironment, able to sustain myeloma cell growth, is present. Two types of syngeneic models have been established: the first one consists in the transplant of murine myeloma cells into other mice (5TMM), while the second one (Vκ*MYC mouse) is a genetically engineered mouse where MYC is activated in germinal center B-cells via an activation-induced cytidine deaminase (AID)-dependent mechanism. SCID, severe combined immune deficiency.
CLINICAL AND LABORATORY FEATURES
Table 107–1 summarizes the signs and symptoms associated with myeloma. The International Myeloma Working Group has issued simplified criteria for the classification of myeloma and related disorders.231,232 Symptomatic myeloma is diagnosed by evidence of organ or tissue impairment (end-organ damage) manifested by anemia, hypercalcemia, lytic bone lesions, renal insufficiency, hyperviscosity, amyloidosis, or recurrent infections, (commonly referred to as the “CRAB” criteria; namely, hypercalcemia, renal failure, anemia, and bone lesions) (Tables 107–2 and 107–3; Fig. 107–6). Presence of these features necessitate immediate treatment for myeloma.
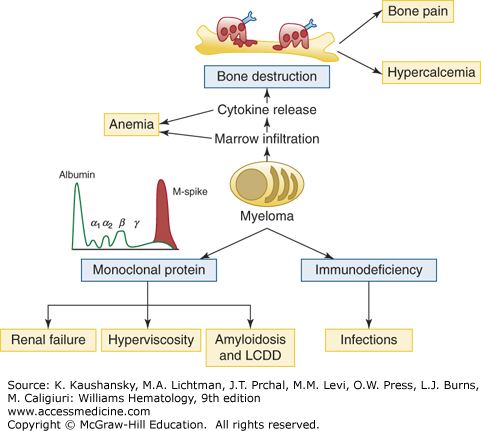
Figure 107–6.
Summary of clinical manifestations of myeloma. Bone destruction, immunodeficiency, and presence of a monoclonal protein account for the main factors capable of inducing symptoms in myeloma patients. Anemia, hypercalcemia, bone pain, and renal failure represent the classical symptoms of myeloma presentation, together with increased risk of infections. Hyperviscosity and amyloid light-chain amyloidosis or light-chain deposition disease (LCDD) are less common presenting situations.
|
| Symptoms and Laboratory Features | Frequency (%) |
|---|---|
| Bone pain (spine, chest, less common in long bones) | 58 |
| Weakness and fatigue | 32 |
| Anemia | 73 |
| Elevated creatinine | 48 |
| Hypercalcemia | 28 |
| Serum monoclonal immunoglobulin (Ig) peak on standard electrophoresis | 82 |
| Weight loss | 24 (one-half of whom had lost ≥9 kg) |
| Monoclonal Ig peak on immunofixation of serum or urine | 97 |
| Monoclonal IgG | 52 |
| Monoclonal IgA | 21 |
| Monoclonal light chains only | 16 |
| Monoclonal IgD | 2 |
| Biclonal | 2 |
| Monoclonal IgM | 0.5 |
| Negative | 6.5 |
| Urinary monoclonal light chains | 75 |
| Marrow plasmacytosis >10% | 90 |
Myelomatous involvement of the marrow typically causes anemia, which is present in more than two-thirds of patients with myeloma and relates to the degree of marrow infiltration. The erythropoietin response is insufficient in myeloma, owing to production of cytokines, (IL-1, TNF-β, Fas ligand, MIP-1α, and tumor necrosis factor–related apoptosis-inducing ligand [TRAIL], which produce erythroblast apoptosis),233 increased serum viscosity, or concomitant renal dysfunction.234,235,236 Patients with myeloma have high urinary and serum hepcidin levels that inversely correlate with hemoglobin values.237,238 IL-6 and bone morphogenetic protein (BMP)-2 mediate hepcidin transcription in the liver via STAT3 signaling,239 which, in turn, blocks iron release from macrophages and inhibits iron absorption from the intestine.240,241 In contrast to other lymphoproliferative disorders, thrombocytopenia is uncommon at diagnosis, even with extensive marrow infiltration, as IL-6 has thrombopoietic activity.242 In some patients, thrombocytopenia might occur secondary to treatment or to autoimmune mechanisms (such as those accounting for anemia or factor VIII deficiency46,243,244,245). Specifically, bortezomib causes a cyclic thrombocytopenia, with a different kinetic from cytotoxic drugs, appearing during the first 10 days of each cycle, but with a short recovery time, no cumulative or persistent effects and absence of marrow megakaryocyte toxic damage, as it primarily results from a functional alteration in platelet budding.246 Prolonged exposure to alkylating agents can also promote onset of a concomitant myelodysplastic syndrome. Overt bleeding is a relatively uncommon presenting symptom for myeloma patients247; however it occurs more commonly with IgA paraproteins, in the presence of very high concentrations of serum immunoglobins, high serum viscosity, and a prolonged bleeding time, with normal platelet counts, prothrombin time (PT), activated partial thromboplastin time (aPTT), and thrombin time.248,249 Acquired von Willebrand factor (VWF) deficiency can develop250 as a result of plasma VWF-neutralizing antibodies, antibodies binding to the VWF glycoprotein 1b binding domain, or interfering with VWF binding to collagen,251,252 or immunoglobulins that nonspecifically coat platelets or bind to fibrin, preventing aggregation. An asymptomatic prolonged thrombin time can also be present,253 as a result of interference with fibrin clot formation by the monoclonal protein254; in few cases, paraproteins recognizing thrombin and factor VIII have been reported.253,255 Bleeding may also result from progression of disease, renal insufficiency, infections, therapy-related toxicity, invasive procedures, and anoxia/thrombosis in the capillary circulation. Bleeding is more common in systemic AL amyloidosis (15 to 41 percent of patients at diagnosis),256,257,258 because of the deposition of free immunoglobin light chains forming insoluble fibrils in the small vessels or, more rarely, as a result of acquired factor X deficiency, related to absorption of factor X onto AL fibrils in the liver and spleen.259,260 Amyloid splenic infiltration can cause hyposplenism and thrombocytosis.260 An increased risk of venous thromboembolic events is typical of MG and myeloma patients.261,262 Hypercoagulable states may result from the pro-inflammatory activity of IL-6, abnormal interactions between myeloma cells, BMSCs and endothelial cells,263 paraprotein effects on fibrin polymerization and resistance to fibrinolysis, or rarely neutralizing antibodies against protein C, S,264,265 or lupus anticoagulant266 and acquired protein C resistance.267 IMiDs, such as thalidomide and lenalidomide, have anti-angiogenic properties and are associated with an increased risk of venous thromboembolism (VTE), ranging from 5 percent268 to 18 percent of treated patients,269 especially when combined with high-dose dexamethasone, doxorubicin, or erythropoietic agents.270,271,272,273,274 Warfarin and low-molecular-weight heparin play a role in primary and secondary VTE prevention,275 but aspirin is normally used in patients treated with IMiDs and steroids, in view of the presence of increased aggregation between platelet and VWF antigens.276,277,278,279
The majority of myeloma patients produce and secrete a monoclonal immunoglobulin (M-protein or M-spike) that can be detected by protein electrophoresis of the serum (SPEP) and/or of urine (UPEP) after a 24-hour urine collection. The M-protein presents as a single narrow peak, migrating in the γ, or rarely β, region of the densitometer tracing. Myeloma cells can secrete immunoglobulin heavy chains plus light chains, light chains alone, or neither (nonsecretory myeloma). In this last case, cytoplasmic immunoglobulins are detected. Immunofixation analysis identifies the unique and specific immunoglobulin idiotypes.47 Monoclonal IgG (usually >3.5 g/dL) is present in approximately 60 percent of myeloma patients, monoclonal IgA (typically >2 g/dL) in 20 percent of patients, monoclonal immunoglobulin light chains alone are detected in 20 percent of patients, while IgD, IgM, and biclonal myeloma are rare (5 percent of cases). A low M-spike concentration is particularly suggestive of IgD myeloma isotype. Light-chain myeloma patients should be followed by UPEP and urine immunofixation and present more often with renal failure or increased creatinine levels. Light-chain proteinuria is frequent especially in IgD myeloma (see Table 107–3). Traditionally, IgA and especially IgD isotypes have been considered prognostically unfavorable.280,281 Their prognostic value has been confirmed in patients enrolled on Total Therapy 1, 2, and 3 protocols; IgD was of borderline significance, mainly because of its rarity, but was strongly associated with elevated β2-microglobulin (β2M) and lactic dehydrogenase (LDH) serum levels, reflecting high tumor burden.282 Suppression of normal, polyclonal serum immunoglobulins resulting in total hypoglobulinemia and an increased risk of infection is present in 70 to 90 percent of patients. The κ light-chain isotype is twice as common as the λ isotype, except in IgD myeloma. The free light chain (FLC) assay (FREELITE assay) is a novel technique used to detect monoclonal FLCs, and provides a FLC κ:λ ratio (Fig. 107–7).283 The κ:λ ratio is considered abnormal if less than 0.26 (λ-restricted Ig) or more than 1.65 (κ-restricted Ig).284 Because the half-life of FLCs is only 2 to 4 hours, in contrast to the half-life of the entire immunoglobulin, which is 17 to 21 days, the FLC assay can be used to detect early treatment responses and should be evaluated routinely in patients with AL amyloidosis and oligosecretory myeloma.285 A normal FLC ratio is also included in the criteria for stringent complete response (sCR).8 The FLC ratio is considered a predictor of the risk of progression from MG or smoldering myeloma to active myeloma286,287 and it has prognostic value, being related to tumor burden.285 Indeed, high baseline FLC correlates with shorter survival in newly diagnosed myeloma patients despite achievement of complete response.288,289 A rapid reduction in FLCs after therapy is also linked to inferior overall and event-free survival, suggesting the presence of highly proliferative myeloma cells, particularly sensitive to combination chemotherapy.290
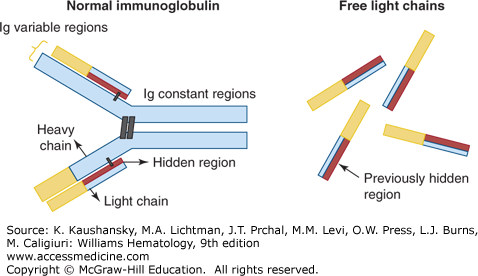
Figure 107–7.
Free light-chain assay description. Normal immunoglobulins are composed of two heavy chains and two light chains, which together form a constant region and a variable region, capable of recognizing specific antigens. The free light-chain assay is used to quantify the amount of free light chains in myeloma patients and to specifically recognize a “hidden” antigenic region (in red) that is normally not detectable from intact immunoglobulins.
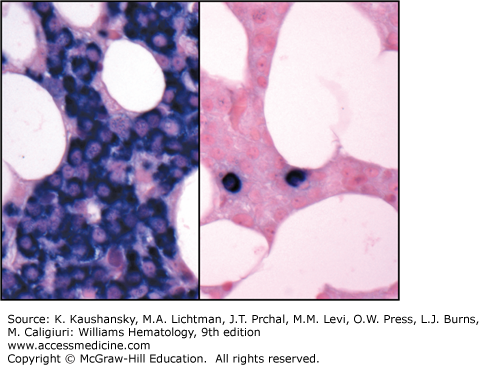
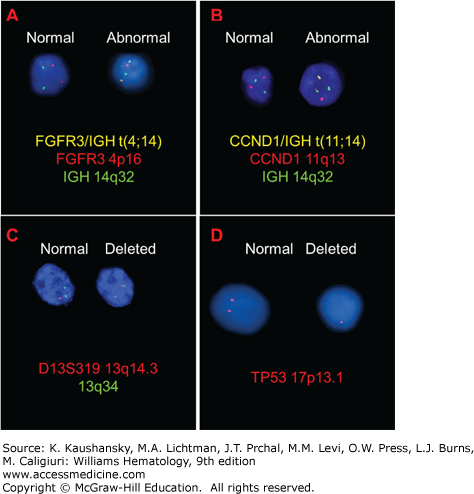
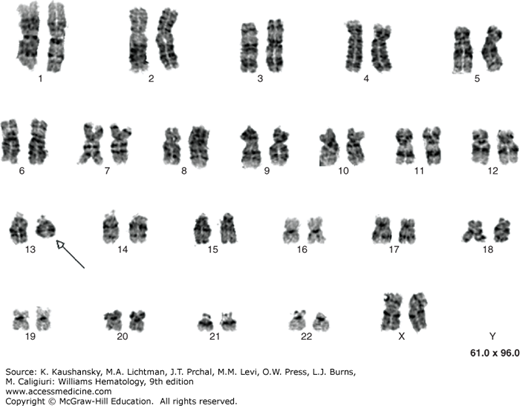
A marrow aspirate or biopsy showing plasmacytosis is a key component for the diagnosis of myeloma. The marrow can be evenly infiltrated (diffuse involvement) but more commonly displays considerable site-to-site variability (focal involvement).291 The percentage of plasma cells can range from 10 percent to a virtual complete replacement of marrow. Occasionally, the biopsy specimen may contain a normal proportion of plasma cells as a result of the patchy marrow involvement. Myeloma diagnosis is also considered in the presence of less than 10 percent of chain restricted plasma cells if symptoms are reported or after histopathologic confirmation of an intraosseous or extraosseous plasmacytoma. By light microscopy, the morphologic appearance of myeloma plasma cells can be indistinguishable from normal plasma cells, characterized by abundant basophilic cytoplasm and round, eccentrically located nuclei, with “clock-face” or “spoke-wheel” chromatin without nucleoli, or by bizarre plasma cells with giant cellular size, open chromatin, and punched nucleoli (indicating increased transcriptional activity), a high frequency of binucleate or multinucleate cells, and the presence of inclusion globules of condensed or crystallized cytoplasmic immunoglobulin, including Russell bodies (cherry-red refractive round bodies), multiple pale bluish-white, grape-like accumulations (Mott cells or Morula cells), crystalline rods, glycogen-rich IgA (flame cells), or other inclusions. These abnormal cells are characteristic of plasmablastic myeloma,292,293 a poor prognostic type of myeloma with a high number of mitotic figures (Figs. 107–8,107–9,107–10).294 Myeloma cells are clonal by definition and produce either κ or λ light chains, which are present in the cytoplasm but not on the membrane surface. A κ:λ ratio greater than 4:1 (normal 2:1295) or less than 1:2 is considered an index of κ or λ monoclonality, distinguishing this condition from reactive plasmacytosis.296 Two-parameter flow cytometry staining for nuclear DNA content by propidium iodide and anti-κ and anti-λ light-chains can also be used to quantitate marrow involvement (Fig. 107–11).297 Myeloma cells are normally CD138+, CD45–, CD38+, and CD19–298, and are CD56+ in 70 percent of patients.299,300,301 A few cases are CD20+302 or CD117+ (KIT),303 but responses to treatment with rituximab or imatinib mesylate are uncommon. If amyloid deposition is suspected, Congo red staining can be performed on the marrow biopsy, showing diffuse involvement or focal perivascular niche localization of amyloid protein. Microvessel density can be assessed by staining for endothelial markers such as CD131 and CD34 in specialized laboratories or during clinical trials.304 Secondary myelodysplastic changes can rarely develop after prolonged treatment in myeloma patients, presenting with pancytopenia in the context of a hypercellular marrow, with characteristic FISH abnormalities (Chap. 87).305,306 Metaphase cytogenetic studies and interphase FISH analysis should be performed routinely on myeloma cells at diagnosis to evaluate the presence of abnormal karyotypes and poor prognostic chromosomal abnormalities, such as deletion of chromosome 17, gain of chromosome 1q, loss of chromosome 1p, deletion of chromosome 13, and t(4;14) or t(14;16) translocations (Figs. 107–12 and 107–13).63,307,308,309,310 Genetic analysis should be repeated at relapse only in patients initially classified as genetic standard-risk to rule out emergence of a more aggressive clone.311 The plasma cell labeling index corresponds to the percentage of plasma cells in the S-phase of the cell cycle and is measured by immunofluorescence staining using an antibody against 5-bromo-2′-deoxyuridine, which is actively incorporated by DNA on marrow plasma cells. Actively cycling myeloma cells represent a small proportion of the total malignant cells, normally 0.5 percent on average,312,313,314,315,316,317 with few patients with a labeling index of more than 5 percent.318,319 This value has been proposed as a myeloma prognostic marker, as patients with a labeling index of more than 0.5 percent at diagnosis have a shorter event-free survival (EFS) and OS.318
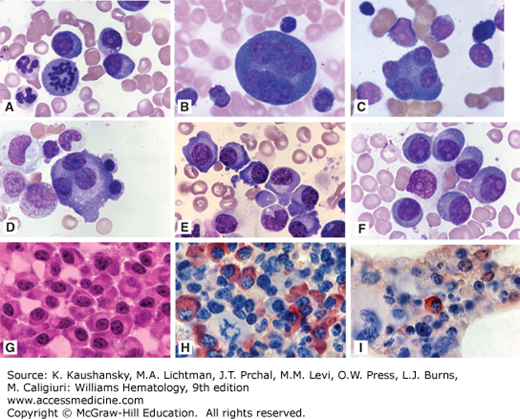
Figure 107–8.
Marrow findings in myeloma. A. Marrow film. Infiltrate of neoplastic plasma cells (myeloma cells). These cells resemble normal plasma cells in their appearance with characteristic nuclear: cytoplasmic area ratio, blocky nuclear chromatin pattern, intense cytoplasmic basophilia, and a prominent “hof” or clear area (Golgi zone). CD138 immunohistochemistry staining is shown in the small box. B. Marrow film. Infiltrate of malignant plasma (myeloma) cells. C(A). Marrow section. Mature malignant plasma cells. C(B). Marrow film. Malignant plasma cells showing prominent perinuclear Golgi apparatus (Hof). C(C) and C(D). Immature plasma cells with abnormal nuclei and size and aberrant morphology.
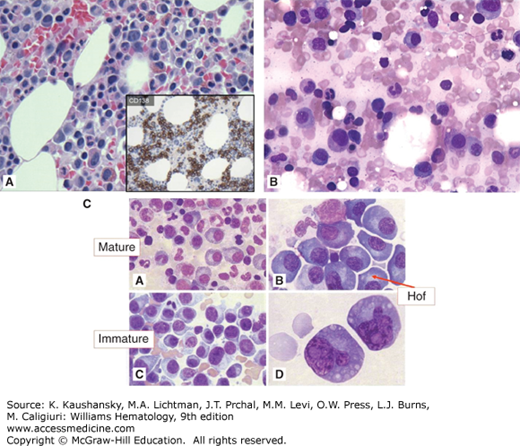
Figure 107–9.
Myeloma: morphologic appearances. A. Marrow film showing replacement by malignant plasma (myeloma) cells. Note classical oval cell shape, eccentric nucleus, striking paranuclear clear area, deeply blue cytoplasm. B. Marrow biopsy section showing replacement by myeloma cells. C. Blood film in a patient with plasma cell leukemia. Three myeloma cells in the blood film field. D and E. Marrow films. Flaming-type giant myeloma cells. Reddish peripheral cytoplasmic coloration reflecting very high concentration of carbohydrate, characteristic of IgA myeloma. The peripheral cytoplasm contains numerous dilated cisterns of the endoplasmic reticulum distended with immunoglobulin. Flaming plasma cells may occasionally be found in IgG myeloma and in reactive plasmacytosis. F. Morula or Mott cell. Myeloma cell engorged with globules presumably containing immunoglobulin. These globules individually are referred to as Russell bodies and plasma cells may be found containing one, several, or many such bodies. G. Plasma cell with immunoglobulins containing globules overlying the nucleus but presumably cytoplasmic in location along with smaller cytoplasmic globular inclusions. H. Immunoglobulin crystal with several globules of immunoglobulin on either side. Note remarkable distortion of the cell to accommodate the crystal. I. Marrow film. Myeloma cells exhibiting cytoplasmic shedding. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accesmedicine.com.)
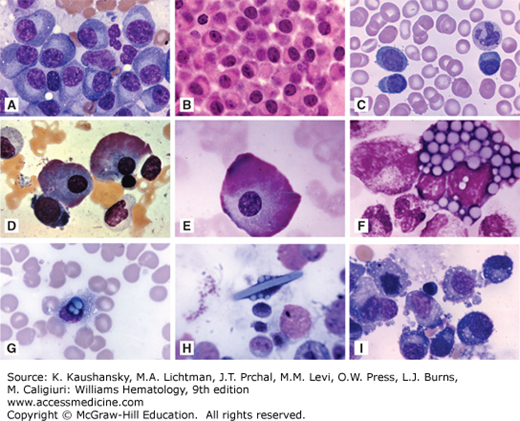
Figure 107–10.
Myeloma: Additional morphologic appearances. A. Marrow film. Three characteristic malignant plasma (myeloma) cells and one in mitosis. B. Marrow film. Giant multinucleated myeloma cell. C. Marrow film. Tetranucleated myeloma cell. D. Marrow film. Trinucleated myeloma cell. E. Marrow film. Infiltrate of classical well-differentiated myeloma cells (plasma cell phenocopies). Eccentric nuclei, paranuclear clear zone, deeply blue (basophilic) peripheral cytoplasm. F. Infiltrate of immature myeloma cells with more circular than ovoid shapes, very large, prominent large nucleoli, less-intense basophilic cytoplasm, less-discrete paranuclear clear zone (plasmablasts). G to I. Marrow biopsy sections showing striking infiltrate of myeloma cells. G. Hematoxylin-and-eosin stain. H. Immunostained for κ light chains, showing frequently positive cells evident by deep rust color in cytoplasm. I. Immunostained for λ light chains showing negative reaction with rare positive cell. Approximately 20:1 κ:λ ratio. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accesmedicine.com.)
Increased creatinine levels (>1.5 to 2.0 mg/dL) occur in 30 to 50 percent of myeloma patients at diagnosis, while overt renal failure requiring hemodialysis affects up to 10 percent of patients.247 Renal insufficiency is related to two major causes: myeloma cast nephropathy (also called light-chain cast nephropathy or myeloma kidney) and hypercalcemia.320 In myeloma cast nephropathy, the tubular absorptive capacity for light chains is overwhelmed, leading to the formation in the distal convoluted tubule (DCT) of the nephron of tubular casts. These tubular casts derive from the binding of precipitated light chains to Tamm-Horsfall mucoprotein (uromodulin) and can obstruct the DCT and parts of the ascending loop of Henle, initiating a giant cell reaction which leads to interstitial inflammation and fibrosis (interstitial nephritis).321 The cast formation rate is strongly related to the urinary free-light chain concentration, which can be estimated by the amount of total proteinuria, based on the 24-hour urine collection or the serum light chain values. Conversely, the urine dipstick may be negative for protein as immunoglobulin light chains are often not detected by this technique. Lambda light chains tend to be more nephrotoxic than the κ type and renal impairment can be present with minimal λ light-chain secretion. Hypercalcemia (calcium >11 mg/dL), the second cause of nephropathy, is present in 15 percent of patients at diagnosis. Hypercalcemia creates volume depletion, natriuresis, and renal vasoconstriction, with an increased risk of prerenal azotemia; moreover, it can lead to intratubular calcium deposition, increasing the toxicity of filtered light chains or cause a reversible form of nephrogenic diabetes insipidus.322 Light-chain glomerulopathy is caused by the deposition of immunoglobulins either in the form of amyloid or nonamyloid. In AL amyloidosis, light-chain immunoglobulin proteinuria is associated with glomerular damage, resulting into an overt nephrotic syndrome (Chap. 108).323 Light chains are converted into insoluble fibrils or granular deposits inside the mesangial cells causing Congo red-positive amyloid accumulation, localized predominantly in the glomeruli. Vascular and tubular amyloid deposits are less common, but can cause narrowing of the vascular lumens or tubular dysfunction such as type 1 (distal) renal tubular acidosis or nephrogenic diabetes insipidus.324 AL amyloidosis with renal dysfunction is more common in patients with λ light-chains, especially those with λ VI light-chain subgroup.325 Kappa chains or heavy-chain fragments can form Congo red-negative nonfibrillar deposits with linear involvement of the basement membrane, in a condition called LCDD or, more generally, monoclonal immunoglobulin deposition disease (MIDD). The clinical presentation of LCDD is heralded by development of the nephrotic syndrome followed by renal failure or acquired Fanconi syndrome (more often associated with κ light-chain deposition). In these deposition diseases, the urine dipstick for protein is positive, as a result of the glomerular leakage of albumin.256,324 Renal enlargement can be caused by AL amyloidosis (Chap. 108) or, less commonly, by renal plasmacytomas.326 Renal vein thrombosis, hyperviscosity, dehydration, use of nephrotoxic drugs (antibiotics, nonsteroidal antiinflammatory drugs, imaging contrast agents—especially when rapidly infused),327 hyperuricemia, or type I cryoglobulinemia can all induce or aggravate renal impairment in myeloma patients. Bisphosphonates, in particular, should be infused slowly, at adjusted doses, based on creatinine values. Renal biopsy is usually unnecessary, unless nephrotic syndrome is present. However, if systemic amyloidosis or, less likely, LCDD, is suspected, a subcutaneous fat aspirate or rectal biopsy should be performed first and tested for amyloid deposits.320 Renal biopsy specimens should be processed fresh-frozen to allow for immunofixation studies, including electron microscopy and Congo red staining for amyloidosis. Supportive care associated with prompt initiation of antimyeloma treatment is the cornerstone of the management of renal impairment in myeloma. To correct hypercalcemia, aggressive hydration, use of calcitonin and a slow infusion of one single dose of a bisphosphonate is applied. Cytoreductive chemotherapy should be started as soon as possible. Rapid removal of light chains by plasma exchange is a controversial technique; the introduction of high cut-off hemodialysis, which employs novel dialysis filters capable of clearing away free-light chains, is showing promising results and improved patient outcomes.328,329,330
In general, myeloma renal impairment is reversible in approximately 50 percent of patients. Conversely, amyloid- and LCDD-related renal impairment tends to be stable or progressive. Patients presenting in acute renal failure have a high early mortality, with up to 30 percent dying within the first months. Improvements in renal function rarely occur after 6 months from diagnosis. Renal dysfunction is a negative prognostic factor, results in use of suboptimal therapies, longer hospitalization, and an increased risk of infection. Hence, patients who recover normal kidney function have a better outcome compared to those who do not.331,332,333
Back or chest bone pain as result of vertebral or rib fractures at sites of osteopenia or from lytic bone lesions is present at the time of diagnosis in approximately 60 percent of patients.247 The pain is usually worse with movement and at night. Pathologic fractures of long bones can ensue as well. Kyphosis or reduction of patient’s height is another common feature. Localized pain can also derive from focal plasmacytomas, presenting as expanding masses compressing the spinal cord or nerve roots. Amyloid deposits can provoked painful mass effects, when localizing into nerve sheaths, as in amyloid-associated carpal tunnel syndrome.334
Myeloma patients are at an increased risk for infections that represent a leading cause of morbidity and mortality. Several aspects contribute to infection risk, including immune dysfunction in the innate and adaptive immune systems,335 extrinsic factors, like type and duration of therapy (e.g., cytotoxic agents, glucocorticoids, lenalidomide, autologous/allogeneic hematopoietic stem cell transplantation), and physical factors, such as age, coexisting comorbidities, hypoventilation secondary to pathologic fractures, indwelling vascular catheters and impaired mucosal integrity. A broad immune dysfunction involving B lymphocytes, T lymphocytes, NK cells, and dendritic cells is noted in myeloma patients.335,336,337 Specifically, myeloma cells or BMSCs can produce a series of immunologically molecules, such as TGF-β, IL-10, and IL-6. TGF-β and IL-10 suppress IL-2 autocrine pathways, blocking T-cell antitumoral responses338,339,340 and stimulate T-regulatory cell proliferation.341 Moreover, these cytokines sustain the immature phenotype of dendritic cells, highly specialized antigen-presenting cells (Chap. 21), reducing their expression of costimulatory molecules (HLA-DR, CD40, and CD80 antigens) and thereby their antigen stimulatory capacity.342 IL-6 also inhibits dendritic cell production from CD34+ progenitor cells.342 Normal CD19+ B lymphocytes at early and late stages are suppressed in myeloma, resulting in hypogammaglobulinemia, inversely correlating with the disease stage.343 B-cell dysfunction in myeloma can be related to TGF-β effects, lack of stimulatory signals from helper T cells, and altered gene expression. Hypogammaglobulinemia is particularly responsible for a myeloma patients’ susceptibility to encapsulated organisms, such as Streptococcus pneumoniae and Haemophilus influenzae. T-cell subsets are also abnormal, with inversion of the CD4:CD8 ratio344 and T-helper type 2 (Th2) cell–type skewing.345 Moreover, global T-cell receptor (TCR) diversity is reduced, with the presence of oligoclonal expansions of CD4+ and CD8+ T cells and TCR signaling is compromised344,346,347; χδT cells and NK cells are also aberrant in myeloma. The presence of decreased B cell or T cell counts can negatively impact survival.348 β2M is the invariant chain of the major histocompatibility (MHC) class 1 molecule, which is shed by myeloma cells. Its levels correlates with tumor burden and are important for patient staging using the International Staging System (ISS)232 (see section Staging below). β2M induces IL-6, IL-8, and IL-10 production and activation of STAT3. However, it also has immunosuppressive functions, by reducing surface expression of CD83, HLA-ABC, costimulatory molecules, and adhesion molecules on dendritic cells, impairing stimulatory dendritic-allospecific T-cell responses by inactivation of Raf/MEK/ERK cascade and NF-κB in dendritic cells.349 Vaccines, prophylactic antibiotics or antivirals, and intravenous immunoglobulin are preventative measures apt to decrease infections among myeloma patients. Yearly influenza vaccines and a single pneumococcal vaccine at diagnosis is recommended, as myeloma patients can still mount a suboptimal immunologic response. Antiviral prophylaxis (e.g., acyclovir 400 mg twice daily or valacyclovir 500 mg once daily) is mandatory in patients treated with bortezomib combined with glucocorticoid regimens to prevent herpes zoster. Antibiotic prophylaxis is controversial. A study where patients were randomized on a 1:1:1 basis to receive either a daily quinolone, trimethoprim-sulfamethoxazole, or placebo for the first 2 months of treatment did not show a decreased incidence of serious infection (more severe than grade 3 and/or requiring hospitalization) nor of any infection within the first 2 months of treatment or any improvement in response rate or OS.350 However, antibiotic prophylaxis with ciprofloxacin or trimethoprim-sulfamethoxazole might be still beneficial in selected patients, such as those patients with a history of repeated infections, those receiving more intense regimens or those with persistently low CD4+ counts to prevent Pneumocystis carinii pneumonia or other infections. Intravenous immunoglobulin (IVIG) infusion may be considered in patients with recurrent, serious infections despite antibiotic prophylaxis.
Local myeloma growth can cause polyneuropathy by spinal cord or peripheral nerve compression, even though polyneuropathy is not a common presenting symptom, unless in the context of perineuronal or perivascular (vasa nervorum) amyloid deposition.256 POEMS syndrome, or osteosclerotic myeloma, is an exception, where complete polyneuropathy is practically always present along with organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes.351,352 The pathogenesis of POEMS syndrome is largely unknown, but chronic overproduction of proinflammatory cytokines, such as VEGF, plays a major role.
Less than 10 percent of myeloma patients develop signs of hyperviscosity syndrome, a condition that manifests in 10 to 30 percent of patients with Waldenström macroglobulinemia (Chap. 109), because monoclonal IgMs display a higher intrinsic viscosity than other immunoglobulins.353,354,355 Cutaneous or mucosal bleeding is very common in hyperviscosity syndrome, together with blurred vision, headache, vertigo, dizziness, nystagmus, deafness, and ataxia. Circulatory problems, affecting cerebral, pulmonary and renal circulation can rarely ensue in the presence of high blood viscosity (see “Hyperviscosity Syndrome” in Chap. 109).356 Relative serum viscosity but not serum immunoglobulin levels correlates with onset and extent of clinical symptoms. Based on the specific physicochemical properties of classes and subclasses of immunoglobulins (Chap. 75), up to one-quarter of patients with IgA myeloma can have blood hyperviscosity as a result of the tendency of IgA to form dimers or polymers.357 Patients with IgG3 subclass myeloma, whose immunoglobulins have higher propensity to aggregate, can also manifest with hyperviscosity syndrome.358
Plasma cell leukemia (PCL) is diagnosed when more than 2000 myeloma cells/μL are present in the blood or plasmacytosis accounts for greater than 20 percent of the differential white cell count. PCL is rarely manifest at the time of primary presentation and more commonly arises from preexisting myeloma as end-stage disease. In this case, tumor cells have become microenvironment-independent and accumulate in the marrow, but also recirculate in the blood (extramedullary disease).359,360,361,362 However, low levels of circulating plasma cells can be detected in the majority of myeloma patients. By definition, extramedullary disease (EMD) is the presence of a clonal plasmacytic infiltrate outside the marrow. Specifically, it is now considered EMD only if the infiltrate is present at anatomic sites distant from the bone or adjacent soft tissue, hence excluding cases where soft-tissue masses arise in contiguity with the marrow.363 Indeed, in true EMD, plasma cells have an immature, plasmablastic morphology and have a high proliferative index. According to different clinical trials, 6.0 to 7.5 percent of patients screened with magnetic resonance imaging (MRI) or positron emission tomography–computed tomography (PET-CT) have EMD at the time of diagnosis.364,365 Extramedullary masses can localize to several organs, including liver, lymph nodes, spleen, kidneys, breasts, pleura, meninges, and cutaneous sites, and are inevitably associated with elevated levels of LDH366 and a poor response to treatment.367,368,369 Leptomeningeal myelomatosis with abnormal cerebrospinal fluid findings is rare but can manifest at advanced stage.370,371 EMD cells are often CD56– orlow, and present t(4;14) and del(17p) more commonly,372,373 together with TP53 mutations, TP53 nuclear localization,110,374 or high expression levels of focal adhesion kinase (FAK1).375 Additionally, it has been speculated that high-dose melphalan and modern salvage therapies can artificially increase the incidence of EMD as a result of longer duration of treatment, emergence of dormant cells, and poor penetration of the drugs to sanctuary sites like the central nervous system.376
Spinal cord compression can result from an extramedullary plasmacytoma or vertebral fracture and present acutely with severe back pain alongside with weakness or paresthesias of the lower extremities, or bladder/bowel incontinence. It should be considered a medical emergency, evaluated with MRI, and promptly treated with a combination of local radiotherapy, decompressive laminectomy, and systemic chemotherapy to avoid permanent deficits. Specifically, local radiotherapy using less than 30 Gy is potentially curative, in the presence of a solitary plasmacytoma; in patients with systemic disease, the DT-PACE regimen, which combines high-dose dexamethasone pulsing, as part of the combination with oral dexamethasone, daily thalidomide, and 4 days of continuous-infusion cisplatin, doxorubicin, cyclophosphamide, and etoposide, can provide effective treatment and should be followed by local radiation in the absence of symptom relief and lack of tumor shrinkage. If a singular vertebral collapse is evident without plasmacytoma, decompressive laminectomy is the treatment of choice.
INITIAL EVALUATION OF THE PATIENT WITH MYELOMA
Minimal evaluation requirements include a complete blood count with differential white cell count; examination of a blood film for the presence of rouleaux and circulating myeloma cells; a comprehensive serum metabolic panel for the detection of hypercalcemia, renal failure, serum β2M, C-reactive protein, and elevation of LDH (Table 107–4). Myeloma protein studies should include serum protein electrophoresis to quantitate the serum protein electrophoretic pattern in combination with nephelometric quantitation of immunoglobulin levels, serum-free light-chain assay, and a 24-hour urine collection to quantitate 24-hour total urinary protein and determine specific urinary proteins, such as light chains or Bence Jones protein, using urine electrophoresis.
| Complete blood count and differential count; examination of blood film |
| Chemistry screen, including calcium, creatinine, lactate dehydrogenase, BNP, proBNP |
| β2-Microglobulin, C-reactive protein |
| Serum protein electrophoresis, immunofixation, quantification of immunoglobulins, serum-free light chains |
| 24-Hour urine collection for protein electrophoresis, immunofixation, quantification of immunoglobulins, including light chains |
| Marrow aspirate and trephine biopsy with metaphase cytogenetics, FISH, immunophenotyping; gene array, and plasma cell labeling index (if available) |
| Bone survey and MRI; PET-CT (if available) |
| Echocardiogram with assessment of diastolic function and measurement of interventricular septal thickness; EKG (if amyloidosis suspected) |
Immunofixation of serum and urine is needed for the immunoglobulin heavy- and light-chain isotype determination. Serum-free light-chain assay is of particular use in the monitoring of patients with a plasma monoclonal immunoglobulin, the diagnosis and monitoring of patients who would otherwise be considered to have nonsecretory myeloma, and patients who only have light-chain proteinuria. Marrow aspiration and biopsy should include genetic studies (FISH and cytogenetics) and flow cytometry. Newer modalities such as mutational profiling and gene expression studies are being undertaken but are not yet standard of care tests. Radiographic examination usually comprises a metastatic bone survey to detect vertebral compression fractures, osteopenia, and impending fractures of long bones and pelvis. MRI and PET-CT are more sensitive than the bone survey, and better capture early bone disease, the extent of bone disease, and EMD. Both MRI and PET-CT findings have important prognostic implications.209,364 These tests are now being used more frequently specifically in smoldering multiple myeloma (SMM) patients. Assessment of the heart by echocardiogram and electrocardiogram is useful in the right clinical context to detect cardiac amyloidosis and/or LCDD, and, in selected cases, cardiac MRI may be helpful to demonstrate myocardial infiltration. Measurement of brain natriuretic peptide and N-terminal prohormone B-type natriuretic peptide are useful screening tests to detect cardiac dysfunction caused by amyloidosis or LCDD.
Multiple attempts have been made to define clinical and laboratory parameters that have prognostic significance in myeloma.231,377,378 Of the many staging systems, the Salmon-Durie staging system was historically the most commonly used, however, it has been replaced by newer staging systems, which reflect better myeloma biology.379 The Salmon-Durie system relates myeloma cell mass to the extent of bony disease, hemoglobin, and calcium levels, and the monoclonal immunoglobulin levels in serum and urine (Table 107–5). However, measurement of bone disease by skeletal survey in myeloma is observer-dependent and potentially subjective.
|
The ISS is based on two widely available parameters—serum β2M and albumin—and recognizes three stages (Table 107–6). Stage I is defined by β2M less than 3.5 mg/L and albumin 3.5 g or greater/100 mL; stage III is characterized by a β2M of 5.5 mg or greater/L.231 The intermediate stage II has features of neither stage I nor III. β2M correlates with tumor mass and impairment in renal function, whereas a low albumin reflects the effect of IL-6 produced by the microenvironment of myeloma cells on the liver.380,381,382 The different ISS stages were predictive of outcome in an analysis of more than 11,000 patients receiving either standard therapies or melphalan-based high-dose therapy followed by autologous hematopoietic stem cell transplantation (auto-HSCT; Table 107–7). Although predictive of outcome, several shortcomings of the ISS include lack of accounting for cytogenetics and bone disease in patients.
| Study | Regimen | No. of Patients | Cr/nCR (%) | ORR (%) | Outcome |
|---|---|---|---|---|---|
| Rajkumar et al.414 | RD | 223 | 18 | 79 | OS 96% on Rd vs. 87% on RD at 1-year |
| Rd | 222 | 14 | 68 | ||
| Harousseau et al.638 | VAD | 121 | 6.4 | 62.8 | PFS 36 mo Bd vs. 30 mo VAD at 32 mo |
| Bd | 121 | 14.8 | 78.5 | ||
| Reeder et al.639 | CyBorD | 33 | 39 | 88 | N/A |
| Richardson et al.418 | RVD | 66 | 39 | 100 | OS 97% at 18 mo |
| Jakubowiak et al.421 | CRD | 53 | 62 | 98 | PFS 92% at 24 mo |
Imaging studies are an essential part of the diagnosis and management of myeloma. The standard of care in the initial staging of newly diagnosed myeloma is a complete skeletal survey, which includes a posteroanterior view of the chest; anteroposterior and lateral views of the cervical spine, thoracic spine, lumbar spine, humeri, femora, and skull; as well as, an anteroposterior view of the pelvis. Approximately 80 percent of patients with myeloma will have radiologic evidence of bone involvement on skeletal survey. Although widely employed, this modality has limitations. Roentgenographically detectable osteolytic lesions require at least 50 to 70 percent loss of bone mass,383 and hence represent advanced bone destruction. Conventional x-rays have limited sensitivity and, consequently, may miss between 10 and 20 percent of early lytic lesions.384 In addition, reproducibility of skeletal survey results is low and dependent on the expertise of the reveiwer.385 Another limitation of plain x-rays is that they cannot be used to assess response to therapy as lytic lesions seldom show evidence of healing.386 Although skeletal survey remains the gold standard for the initial evaluation of myeloma, there are limitations to this modality that necessitate the use of additional imaging modalities (Fig. 107–14).387
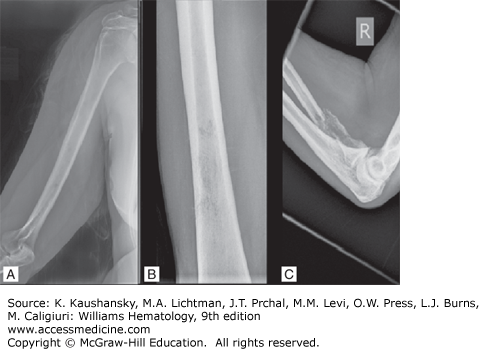
MRI is widely used in both newly diagnosed and relapsed myeloma and in the event of suspected cord compression. Whole-body MRI can give complementary information to skeletal survey and is recommended in patients with normal plain radiography, particularly when symptoms are present. Numerous studies demonstrate superior sensitivity of MRI when compared to both skeletal survey388,389 and whole-body multidetector computed tomography (MDCT).390 MRI also has the ability to discriminate myeloma from normal marrow. Specifically, MRI allows visualization of the medullary cavity and thus direct assessment of the extent of myeloma cell infiltration of the bone.391 In the event of suspected spinal cord compression, MRI is the imaging modality of choice for its ability to provide an assessment of the level and extent of cord compression, the size of the tumor mass, and the extent to which it is compressing the epidural space (Fig. 107–15).392 In the event that MRI is unavailable or contraindicated, urgent CT may be used for the evaluation of a potential cord compression.
Figure 107–15
A. T2-weighted axial imaging of thoracic spine. Myelomatous lesion involves T11 vertebral body and extends into left posterior elements. B. T1-weighted sequencing. Sagittal view of thoracic spine demonstrating diffuse marrow replacement. C. T2-weighted sequencing. Sagittal view of thoracic spine. At T10, there is extraosseous extension of soft tissue within the paravertebral space on the right as well as the ventral epidural space and right lateral epidural space. Soft tissue extends laterally to partly efface the right T10–11 neural foramen.
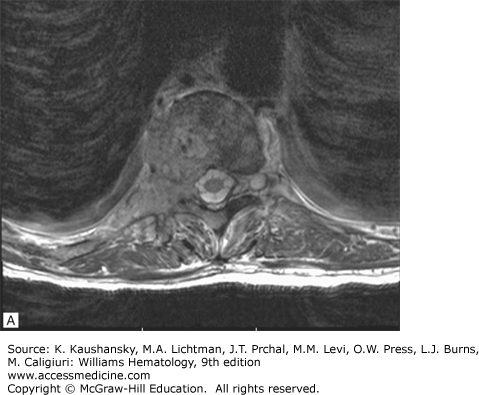
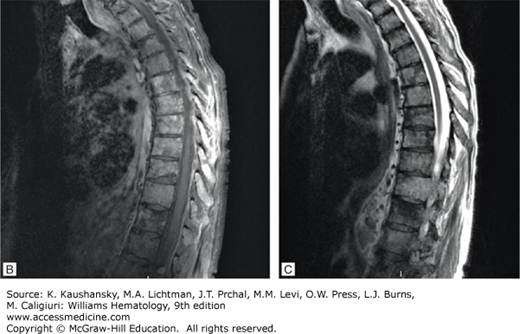
PET, particularly in combination with CT, can be used for the detection of active myeloma. Multiple studies demonstrate that PET-CT is able to detect lesions at least 1 cm in diameter using a standard uptake value (SUV) cutoff of 2.5 to indicate the presence of disease.393 The limitation of this technology is that subcentimeter lesions may not be detected.394 In a prospective study comparing PET-CT, MRI, and whole-body x-rays in newly diagnosed myeloma patients, PET-CT was superior to plain films in 46 percent of patients, including 19 percent with negative x-rays. However, PET-CT scans of the spine and pelvis failed to show abnormalities in 30 percent of patients in whom MRI had demonstrated an abnormal pattern of marrow involvement. On the other hand, PET-CT identified myelomatous lesions in areas that were out of the field of view of MRI in 35 percent of patients. The combination of these two modalities proved the most powerful with a detection rate of as high as 92 percent.395 In a multivariate analysis, the same group also showed that persistent PET-CT positivity before and after primary therapy and subsequent high-dose therapy, is a predictor of prognosis in patients with symptomatic myeloma (Fig. 107–16).396 CT, PET-CT, and MRI can be used for the diagnosis and assessment of soft-tissue masses.
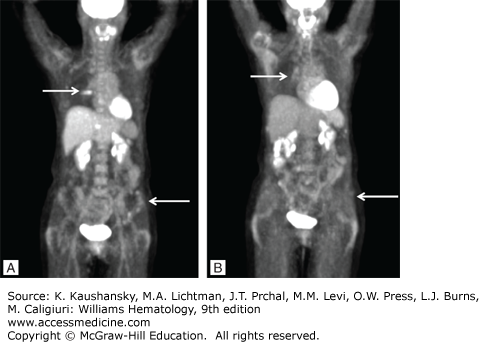
DIFFERENTIAL DIAGNOSIS
If the initial laboratory evaluation indicates the presence of a monoclonal immunoglobulin in serum and/or urine, the finding requires further studies to distinguish among (1) MG; (2) solitary plasmacytoma of bone or soft tissue; (3) indolent myeloma; (4) immunoglobulin deposition diseases, such as primary amyloidosis or LCDD; and (5) symptomatic or progressive myeloma.397 Table 107–5 lists the important studies that can be done to distinguish among these possibilities. Tables 107–1,107–2,107–3,107–4 indicate the findings of symptomatic myeloma. The International Myeloma Working Group has developed new standardized diagnostic criteria that have been widely adopted.398
THERAPY
Myeloma therapy is in a period of dynamic change. The years since 1996 have seen dramatic advances in the treatment of myeloma, beginning with the publication of a randomized trial investigating the use of high-dose melphalan and autologous stem cell transplant in 1996,399
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


