Myelodysplastic Syndromes
INTRODUCTION
Myelodysplastic syndromes (MDS) represent premalignant entities that share many characteristics with acute myeloid leukemia (AML). These clonal hematopoietic stem cell (HSC) disorders are characterized by pancytopenia resulting from failure of normal hematopoiesis. The bone marrow shows hypercellularity, arrested maturation in one or more cellular lineages, and an increase in bone marrow myeloid precursors. Clinical symptoms result from cytopenias. Approximately one-third of patients ultimately progress to AML. Treatment involves supportive care and the use of agents capable of ameliorating cytopenias and delaying development of AML. However, hematopoietic stem cell transplantation (HCT) represents the only potentially curative treatment for MDS.
KEY FEATURES
• One or more peripheral blood cytopenias.
• Hematopoietic cell dysplasia.
• Bone marrow hypercellularity.
• Ringed sideroblasts in a subset of patients.
• Less than 20% bone marrow and peripheral blood myeloblasts.
• Abnormal cytogenetics are observed in approximately 50% of patients. Approximately 20% of patients have characteristic, interstitial deletions within the long arm of chromosome 5, which are associated with clinical response to immunomodulatory drugs (IMIDs).
• MDS may be related to prior chemotherapy and/or radiation for another medical condition (therapy-related MDS or T-MDS).
• Approximately 30% of patients with MDS develop AML.
EPIDEMIOLOGY
Approximately 15,000–30,000 new cases of MDS are diagnosed in the United States each year. MDS is three to four times more prevalent than AML and follows a more indolent course. MDS is likely underdiagnosed. MDS is one cause of anemia in the elderly.
ETIOLOGY
The exact cause of MDS is unknown in most patients. However, intrinsic defects in hematopoietic cells and extrinsic defects associated with the bone marrow microenvironment are involved in the pathogenesis of this disorder.
While most patients with MDS have spontaneously arising, de novo, disease, a portion of patients has therapy-related MDS (T-MDS). Such patients have received chemotherapy and/or radiation in the past, possibly for another malignancy or autoimmune disorder. T-MDS develops within a period of 3–10 years following chemotherapy and is associated with complex chromosomal abnormalities, often involving alterations of chromosomes 5 and/or 7. In addition, T-MDS is associated with a more aggressive clinical course and poorer prognosis in comparison to de novo MDS.
CLINICAL CHARACTERISTICS
• Median age is approximately 70 years.
• Slightly more common in males than in females.
• Prevalence is 50,000–100,000 cases in the United States.
• May be associated with prior chemotherapy, radiation, or environmental exposures to genotoxic agents.
CLASSIFICATION OF MDS
The World Health Organization (WHO) classification system includes refractory anemia (RA), RA with ringed sideroblasts (RARS), refractory cytopenia with multilineage dysplasia (RCMD), and MDS with isolated deletion of 5q for patients harboring a distinct interstitial deletion within the long arm of chromosome 5 (1). Patients with elevated bone marrow blasts of 5%–9% have RA with excess blasts I (RAEB-I) and patients with 10%–19% blasts have RAEB-II. Individuals with 20% or greater myeloblasts in the marrow or peripheral blood have AML.
PROGNOSIS
The most commonly used system to assess a patient’s prognosis is the International Prognostic Scoring System (IPSS) (2). This system assigns a score at diagnosis within each of three categories: the percentage of bone marrow blasts, cytogenetics, and the number and degrees of cytopenias (Table 25-1). The scores are added to yield the IPSS category (Low, Int-1, Int-2, and High). The median survival for patients with Low-, Int-1-, Int-2-, and High-risk disease is 5.7, 3.5, 1.2, and 0.4 years, respectively. Importantly, this system applies only to patients with de novo MDS and was not developed using information from patients with T-MDS, who uniformly have a worse prognosis.
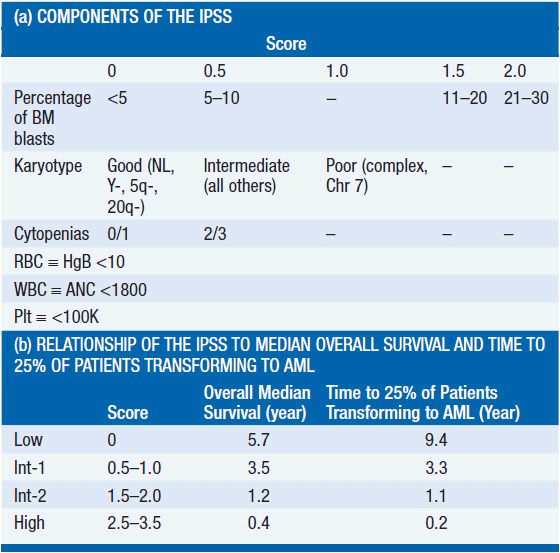
Additional staging systems have emerged. The WPSS system incorporates the WHO category and whether a patient requires transfusions (3). This system may be used dynamically throughout a patient’s illness to assess prognosis. Another system stratifies patients within IPSS categories using five refined cytogenetic subgroups (4). Indeed, a revised IPSS (IPSS-R) has been developed which incorporates these five subgroups along with blast percentage, hemoglobin, platelet count, and ANC (Greenberg P., et al., American Society of Hematology Meeting, December, 2011).
Other factors affect prognosis. The presence of abnormally localized immature progenitors (ALIPS) within the bone marrow is associated with worse prognosis. These collections of immature, CD34 positive cells are displaced from their customary paratrabecular location to the central marrow space and are associated with decreased survival and increased risk of transformation to AML, even within IPSS subgroups. Dependence upon blood transfusions and elevated ferritin levels are also associated with worse prognosis.
KEY ELEMENTS OF PROGNOSIS
• IPSS scoring system (Table 25-1):
• Percentage of bone marrow blasts
• Cytogenetics
• Number and degrees of cytopenias
• Additional parameters important when assessing prognosis:
• ALIPS
• Requirement for blood product transfusions
• Therapy-related MDS
• Elevated ferritin
PATHOPHYSIOLOGY
MDS involvement of multiple hematopoietic lineages suggests the disease arises in a primitive hematopoietic cell, or HSC. However, the microenvironment, too, contributes significantly to disease pathogenesis.
 MDS IS A CLONAL STEM CELL DISORDER
MDS IS A CLONAL STEM CELL DISORDER
The hypercellular bone marrow in MDS is clonal in origin and the clonal genetic lesion resides within a primitive HSC. MDS arises from a primitive, multipotent HSC capable of homing and engraftment.
 GENOMIC INSTABILITY
GENOMIC INSTABILITY
Genomic instability within hematopoietic cells further indicates the presence of a cell-intrinsic defect. Clonal genetic abnormalities are observed in the bone marrow of 50% of patients with de novo MDS and 80% of patients with secondary MDS. The majority of abnormalities are nonrandom. The presence of genetic alterations, in most cases, is associated with inferior prognosis. However, one particular genetic alteration, interstitial deletion within the long arm of chromosome 5, paradoxically confers a favorable prognosis.
T-MDS is an example of the contribution of genomic instability to MDS pathogenesis. T-MDS occurs in younger patients than de novo MDS and is more often associated with chromosomal abnormalities. Karyotypic abnormalities include deletion of large portions of, or entire, chromosomes (-5, -7, 7q-, 13q-, 17p-, and -18) (5).
Clinically, T-MDS follows exposure to agents that cause DNA damage and accounts for approximately 10%–20% of MDS/AML. Affected individuals have been previously treated for lung cancer, breast cancer, childhood acute lymphoblastic leukemia, rheumatoid arthritis, and other oncologic and autoimmune disorders requiring chemotherapy and/or irradiation. T-MDS is associated with a poorer prognosis than de novo MDS, with a median survival of approximately 9 months. Injury associated with topoisomerase inhibitor chemotherapy, such as etoposide, has the earliest onset, often within 2–3 years of exposure. Typical mutations involve core binding factor on chromosomes 16 or 21 and the mixed-lineage leukemia (MLL) gene on 11q. In contrast, alkylating agents such as chlorambucil and cyclophosphamide result in a more latent T-MDS, arising 4–7 years following exposure. Alkylator-associated T-MDS is often associated with abnormalities of chromosomes 5 and/or 7. Exposure to radiation may result in delayed onset of T-MDS, even 10 years or longer following exposure. This category of injury is associated with mutations in the AML1 gene. Genotoxic insult from occupational solvents, such as benzene, is clearly associated with development of MDS/AML. Bone marrow disorders associated with stem cell defects such as paroxysmal nocturnal hemoglobinuria (PNH) and aplastic anemia (AA) may evolve into MDS.
 5Q- INTERSTITIAL DELETION
5Q- INTERSTITIAL DELETION
Approximately 5%–20% of patients with MDS harbor an interstitial deletion within the long arm of chromosome 5, with or without additional cytogenetic abnormalities. This represents the most common single cytogenetic abnormality in MDS (6).
These patients differ from those with T-MDS, who have losses of large regions of 5q or the entire chromosome 5 and have a distinctly inferior prognosis. Instead, interstitial deletion of genomic DNA between bands q13 and q34 is prognostically favorable compared to other types of MDS. Most importantly, patients with 5q- are exquisitely responsive to the class of agents known as immunomodulators (IMIDs). Patients harboring the 5q- interstitial deletion but carrying additional cytogenetic abnormalities are also responsive to IMIDs, although they have a worse prognosis when compared to patients with isolated interstitial deletion of 5q.
Approximately half of patients with 5q- have isolated deletion of 5q- and clinical features comprising the “5q- syndrome”: RA, mild leukopenia, atypical megakaryocytes, normal or increased platelets, transfusion dependence, and extended survival with low risk of transformation to AML. 5q-syndrome is twice as common in women as men and has a median age of 68 years. The pathogenesis and clinical features of the 5q- syndrome may be related to haploinsufficiency of the RPS14 gene and to loss of miR-145, both on chromosome 5q (7, 8).
GENETIC MUTATIONS WITH NORMAL KARYOTYPE
About 50% of patients have somatic mutations in one or more of at least 18 genes. Mutations in RUNX1, TP53, and NRAS are associated with severe thrombocytopenia and a higher percentage of blast cells in the bone marrow (9). Mutations in TP53, EZH2, and ETV6 are associated with a greater than twofold increased risk of death.
ABNORMAL DIFFERENTIATION
A major clinicopathologic feature of MDS is altered differentiation on cytologic examination of the bone marrow aspirate and biopsy, which displays arrested differentiation and dysplasias affecting one or more lineages. In vitro differentiation of MDS bone marrow is diverted toward nonerythroid lineages, explaining the clinical anemia observed in these patients. MDS marrow contains lower levels of erythroid progenitors and requires severalfold higher concentrations of erythropoietin (Epo) to support in vitro erythroid colony growth. This is clinically relevant, as patients with MDS are often resistant to exogenous Epo or require higher doses to support erythropoiesis than required for other diseases.
 INCREASED PROLIFERATION AND APOPTOSIS
INCREASED PROLIFERATION AND APOPTOSIS
Cell cycle analyses have demonstrated increased cellular proliferation in MDS marrow, particularly in the myeloid lineage. However, this is accompanied by an increase in cellular apoptosis. The net balance is a hypercellular bone marrow but ineffective hematopoiesis.
Proliferation and apoptosis have been specifically studied in primitive CD34+ cells from MDS. In early stages of disease such as RA and RARS, apoptosis is greatest and exceeds proliferation. In progressive stages of disease, the ratio of apoptosis to proliferation equalizes. Progression is associated with reductions in both proliferation and apoptosis (10).
 MICROENVIRONMENT
MICROENVIRONMENT
Several abnormalities of the environment in which hematologic progenitors proliferate contribute to the pathogenesis of MDS. The most notable example of the potential role of the microenvironment is evidenced in a murine model where mice lacking expression of Dicer1 strictly within osteoprogenitors develop MDS and, in some case, AML (11).
Another potential contributor to MDS pathogenesis is increased secretion of pro-apoptotic cytokines by bone marrow fibroblasts and macrophages, resulting in increased hematopoietic cell apoptosis. Cells and stroma from MDS patients secrete increased levels of TNF-α, interleukin-6, and IFN-γ relative to normal controls. Agents such as IMIDs inhibit TNF-α and IL-6 secretion and reduce bone marrow angiogenesis.
T-lymphocytes are hypothesized to provide immune surveillance in MDS and undergo activation and proliferation in an attempt to eradicate the malignant clone. Indeed, use of immunosuppressive medications such as cyclosporine and eliminating activated T cells using antithymocyte globulin (ATG) can improve cytopenias, particularly in patients with hypoplastic MDS (12).
CLINICAL PRESENTATION
The clinical presentation of MDS relates to the number and degree of cytopenias. Anemia, the most common cytopenia in MDS and seen in 80%–90% of patients, may manifest as a sensation of light headedness, fatigue, chest pain, dyspnea, palpitations, and depression. Leukopenia is the second most common cytopenia in MDS, present in 50% of affected individuals. Manifestations include recurrent lung, sinus, and skin infections. Thrombocytopenia is present in 25% of patients and may result in easy bruising, epistaxis, petechiae, gastrointestinal bleeding, and hematuria. In addition, qualitative defects in hematopoietic cell function may result in clinical signs and symptoms, even in the presence of adequate blood counts. For example, neutrophil dysfunction may contribute to infection, while platelet dysfunction may result in hemorrhage even if the blood counts are within the normal range.
1. Anemia. Light headedness, fatigue, chest pain, dyspnea, palpitations, and depression.
2. Leukopenia. Lung, sinus, and skin infections.
3. Thrombocytopenia. Easy bruising, epistaxis, petechiae, gastrointestinal bleeding, and hematuria.
DIAGNOSTIC STUDIES
The initial evaluation of uni- or multilineage cytopenias begins with careful history taking. Medications, such as antibiotics and chemotherapeutic agents, are common causes. Also, infections caused by parvovirus, HBV, HCV, EBV, CMV, and HIV suppress hematopoiesis, and their presence may be discerned by history, physical examination, and laboratory studies.
When MDS is suspected, a complete blood count with differential is necessary to identify the number and severity of cytopenias. An elevated erythroid mean corpuscular volume (MCV) is often, although not always, present in MDS. Evaluation of iron levels by assessing the iron (Fe), total iron binding capacity (TIBC), and ferritin levels is necessary to assess baseline iron stores, to determine whether iron supplementation is required to enhance hematopoiesis, or identify situations of Fe overload. The B12 and folate levels should also be assessed to ensure adequate substrates for hematopoiesis. An Epo level is helpful in deciding whether exogenous erythroid growth factor administration will be likely to help patients with anemia. Levels of copper and ceruloplasmin should be checked, particularly in patients with ringed sideroblasts within the bone marrow.
A bone marrow aspiration and core biopsy is essential for the diagnosis of MDS. The percentage of bone marrow cellularity is assessed on the core biopsy, while the myeloblast percentage is calculated using the aspirate. Morphologic assessment of myeloid and erythroid dysplasia is made using the aspirate, while megakaryocyte dysplasia is most easily assessed on the core. Flow cytometry is utilized to identify monotypic populations of lymphoid and myeloid cells. Cytogenetic analysis is performed on the aspirate. If cytogenetic information cannot be obtained, FISH may be used to disclose critical genetic deletions.
Bone marrow studies may reveal alternative causes for cytopenias, such as other hematologic malignancies including AML and non-Hodgkin lymphomas, and solid tumors metastatic to the bone. Hypoplastic MDS, hypoplastic anemia, and aplastic anemia may be determined on the bone marrow core. PNH may be detected by flow cytometric assessment of the glycosylphosphatidylinositol (GPI) linked proteins CD55 and CD59.
Laboratory studies used to diagnose MDS:
• Peripheral blood
• CBC with differential, MCV
• Fe, TIBC, ferritin, copper, ceruloplasmin
• B12, folate
• Epo level
• Bone marrow aspiration and biopsy
• Morphologic analysis of dysplasia using the aspirate and core biopsy
• Quantitation of myeloblasts using the bone marrow aspirate
• Flow cytometry to identify monoclonal populations of lymphoid and myeloid cells, PNH, and T-cell large granular lymphocytes
• Cytogenetics to identify chromosomal alterations (FISH is used when cytogenetic information cannot be obtained)
THERAPIES FOR MDS
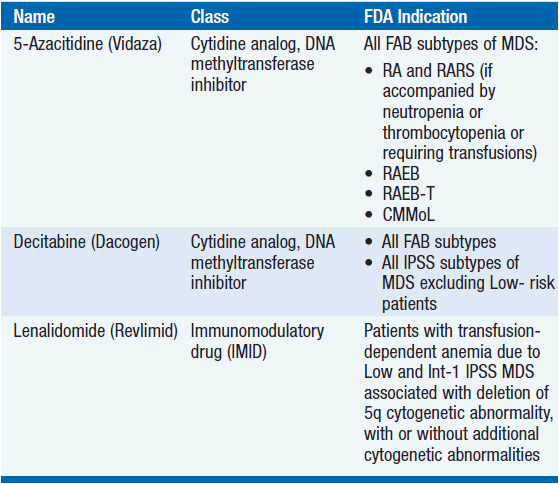
A broad range of management options exists for MDS including amelioration of hematologic deficits with blood product support and administration of growth factors, the use of novel agents aimed at restoring normal hematopoiesis and reducing the malignant clone (Table 25-2), and HCT for patients with high-risk disease for whom a donor can be identified.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


