General Overview
Myelodysplastic syndromes (MDSs) represent a heterogeneous group of myeloid neoplasms characterized by abnormal differentiation and maturation of myeloid cells, ineffective hematopoiesis, and increased risk of transformation to acute myeloid leukemia (AML). Peripheral blood (PB) cytopenias are common, with most patients having multilineage cytopenias.
The diagnosis of MDS requires an integrated approach, including morphologic evaluation of PB and marrow smear and biopsy, immunohistochemistry and flow cytometry (FC), and cytogenetic analyses including standard karyotype and fluorescence in situ hybridization. More recently, molecular tests for mutations and single-nucleotide polymorphisms have become the standard of care as well. MDS is classically a disease of old age. Childhood MDS is not discussed in this chapter.
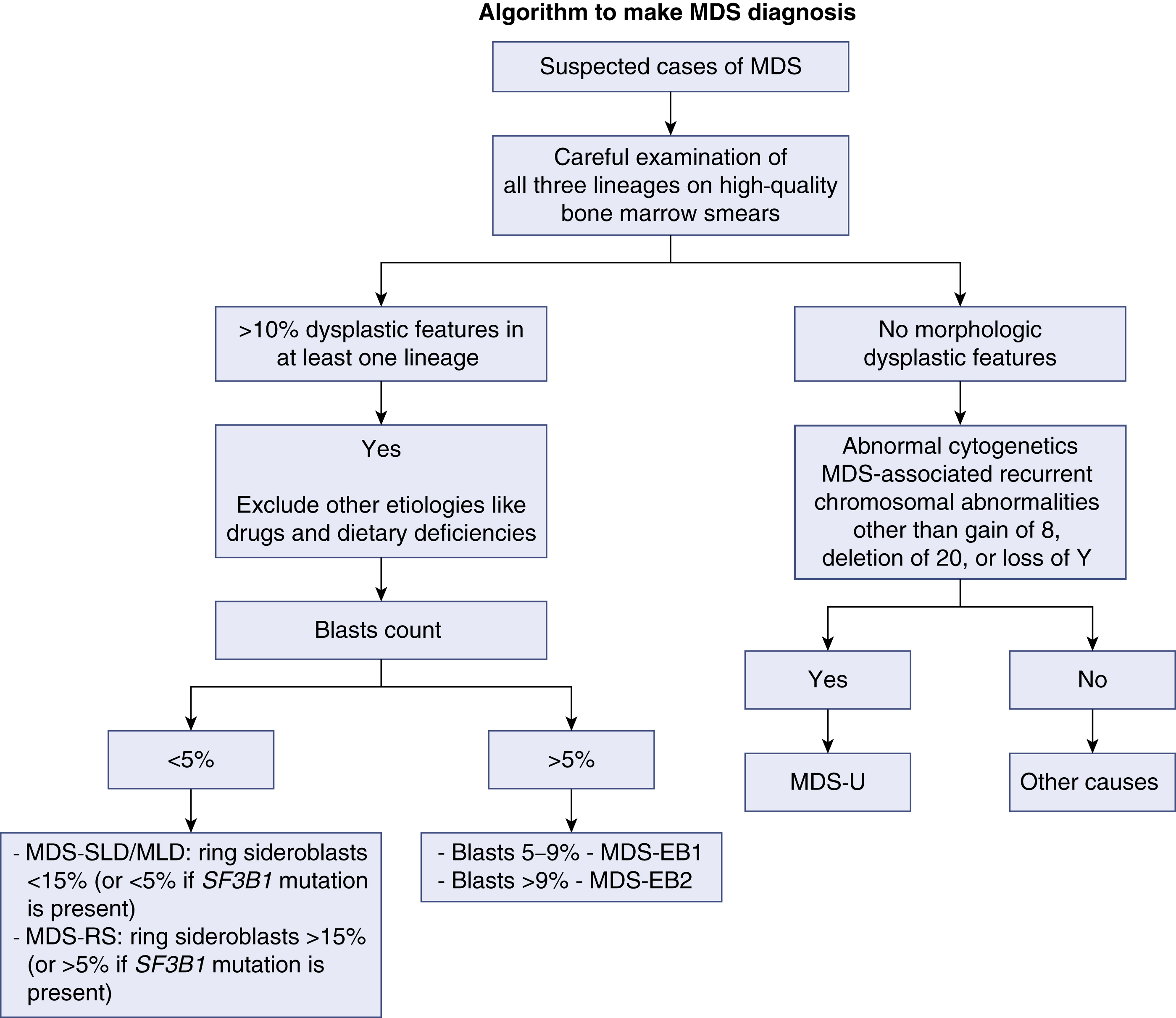
Once the diagnosis of MDS is suspected, the case should be classified accordingly. Using the most recent World Health Organization classification of MDS, classifying any MDS case depends on three criteria: (1) the number of lineages affected by the dysplasia, (2) the presence of ring sideroblasts, and (3) enumeration of blasts. In any case of MDS, the presence of more than 20% blasts in either the bone marrow (BM) or the PB makes the diagnosis of ACL warranted. A proper and complete evaluation of an MDS case requires adequately stained, high-quality BM smears in addition to the BM core biopsy specimen and preferably a PB smear. It is worth noting that MDS with isolated del(5q) is classified in its own category owing to the unique clinical and pathologic features. Morphologic abnormalities and dysplastic features in at least one lineage are the hallmark of diagnosis of any case of MDS. When cytopenias are present without clear morphologic dysplasia, the diagnosis of idiopathic cytopenias of undetermined significance (ICUS) may be considered. Such patients should have close flow-up as they carry a variable risk of developing MDS. The exception to this rule involves patients who show specific cytogenetic abnormalities that are diagnostic of MDS even in the absence of morphologic features. In addition to the WHO classification, there are other schemes developed to confer the prognosis. The International Prognostic Scoring System (IPSS) and the 2012 revised edition (IPSS-R) are the most commonly used. Fig 7.1 shows a schema approach to MDS cases.
The bone marrow (BM) in typical cases of MDS is hypercellular as the result of ineffective hematopoiesis. The myeloid-to-erythroid ratio is usually decreased with a degree of erythroid hyperplasia. The erythroid lineage is the most common affected in MDS. Anemia, which is usually macrocytic, is very common. The ineffective erythropoiesis leads to abnormal iron utilization evident by increase in iron stores and ring sideroblasts. Ring sideroblasts are defined by at least five granules that are positive for Prussian blue stain lining at least one-third of the circumference of the nucleated red blood cell (RBC) precursors. Dysplasia in the erythroid lineage is manifested by a spectrum of abnormalities. Megaloblastoid changes that encompass cytoplasm/nuclear asynchrony and left-shifted maturation in the erythroid lineage are probably the most common. Multinucleation, irregular nuclear contours, and numerous karyorrhectic forms are other features of dyserythropoiesis. It should be noted, however, that those features seen in dyserythropoiesis are not specific to MDS and are commonly seen in reactive conditions that stress the normal erythropoiesis process.
WHO Classification
To address some of the main criticisms of the French-American-British (FAB) classification system, the WHO provided a revised and updated MDS classification system in 2001. The WHO criteria for MDS have been updated twice since then, once in 2008 and more recently in 2016.
The 2016 WHO guidelines for diagnosing MDS are based largely on the degree of dysplasia and the blast percentages. The presence of specific types of cytopenia (e.g., anemia or thrombocytopenia) has less importance in the newest WHO classification system, building on the foundation that the cell lineages exhibiting dysplasia and those exhibiting peripheral cytopenias are not consistently correlated in patients with MDS. , The threshold for dysplasia per WHO guidelines is 10% or more of cells in any lineage, and the guidelines make an effort to acknowledge that secondary causes for dysplasia must be carefully and thoroughly ruled out, as this degree of dysplasia can occur in patients without MDS and a considerable interobserver variation exists when evaluating patients for degree of dysplasia. ,
The current WHO MDS classification is as follows:
- •
MDS with single lineage dysplasia
- •
MDS with multilineage dysplasia
- •
MDS with ring sideroblasts with either single or multilineage dysplasia
- •
MDS with excess blasts 1 (blasts are 5%–9% in the BM or 2%–4% in the PB)
- •
MDS with excess blasts 2 (blasts are 10%–19% in the BM or 5%–19% in the PB)
- •
MDS with isolated del(5q)
- •
MDS unclassified
Morphologic Features
Erythroid Cells
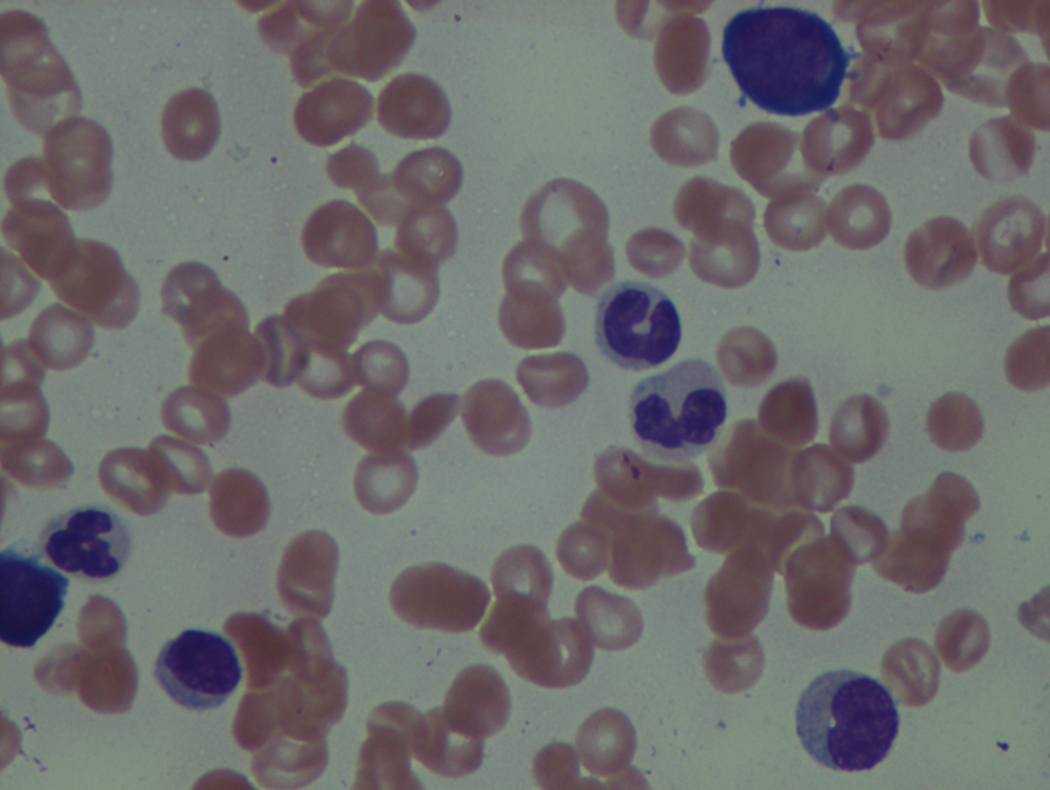
As previously mentioned, anemia is the most common cytopenia seen in MDS, and is present in up to 90% of patients at the time of diagnosis. The anemia in MDS is most often macrocytic, with a low reticulocyte index. There is often anisocytosis with an increased red cell distribution width reflecting the ineffective erythropoiesis. Less commonly, dacrocytes, elliptocytes, or acanthocytes may be observed. , Marked elliptocytosis has been documented to be associated with an acquired deficit in human erythroid protein 4.1, an important structural component of the RBC membrane, seen in MDS with deletion of chromosome 20q. Cases of myelodysplasia usually show decreased myeloid-to-erythroid ratio with erythroid hyperplasia. Figs. 7.2 to 7.14 show the aforementioned erythroid abnormalities.
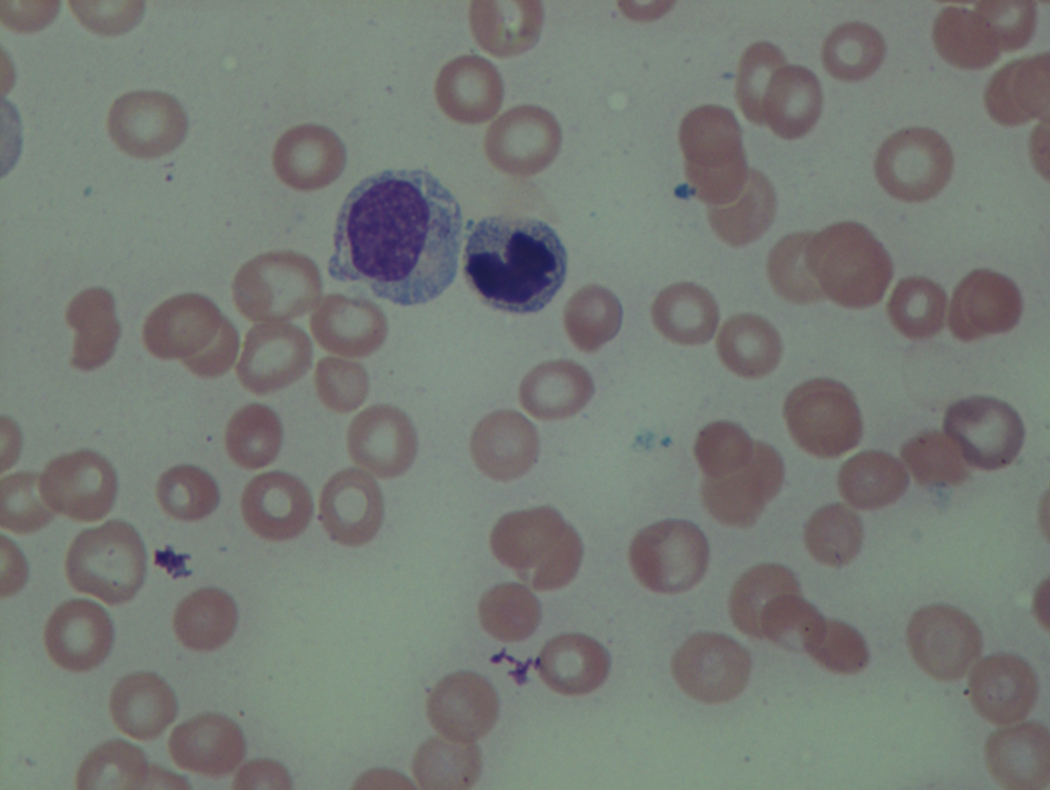
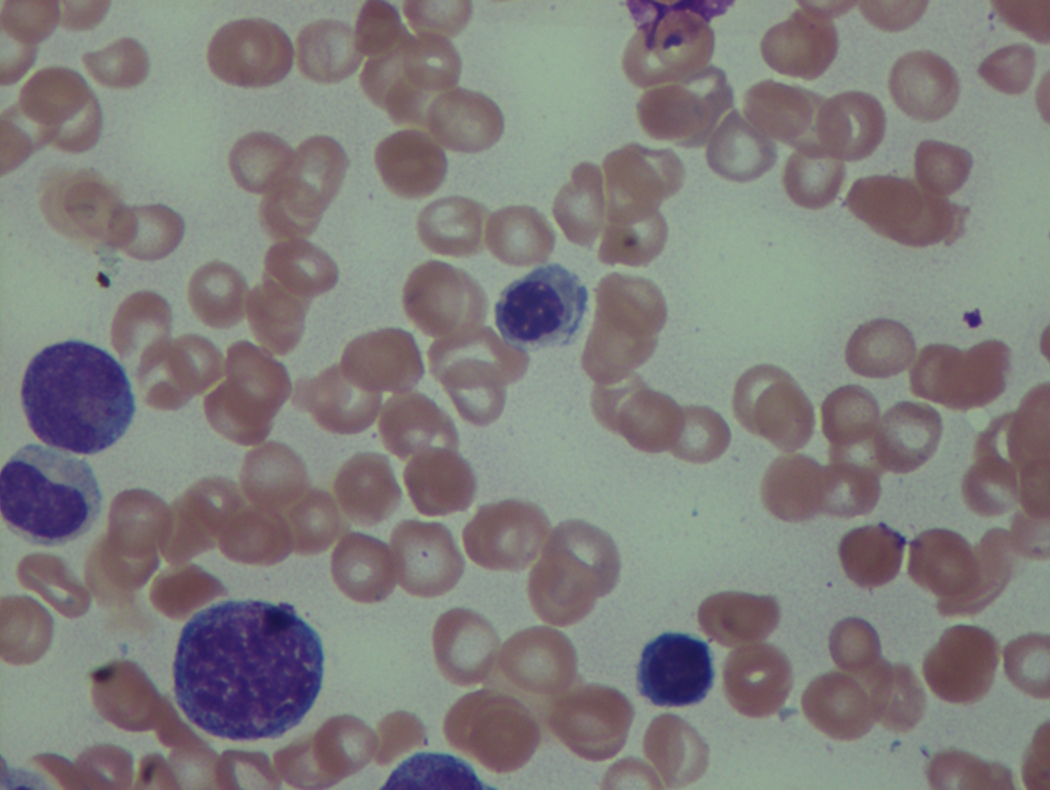
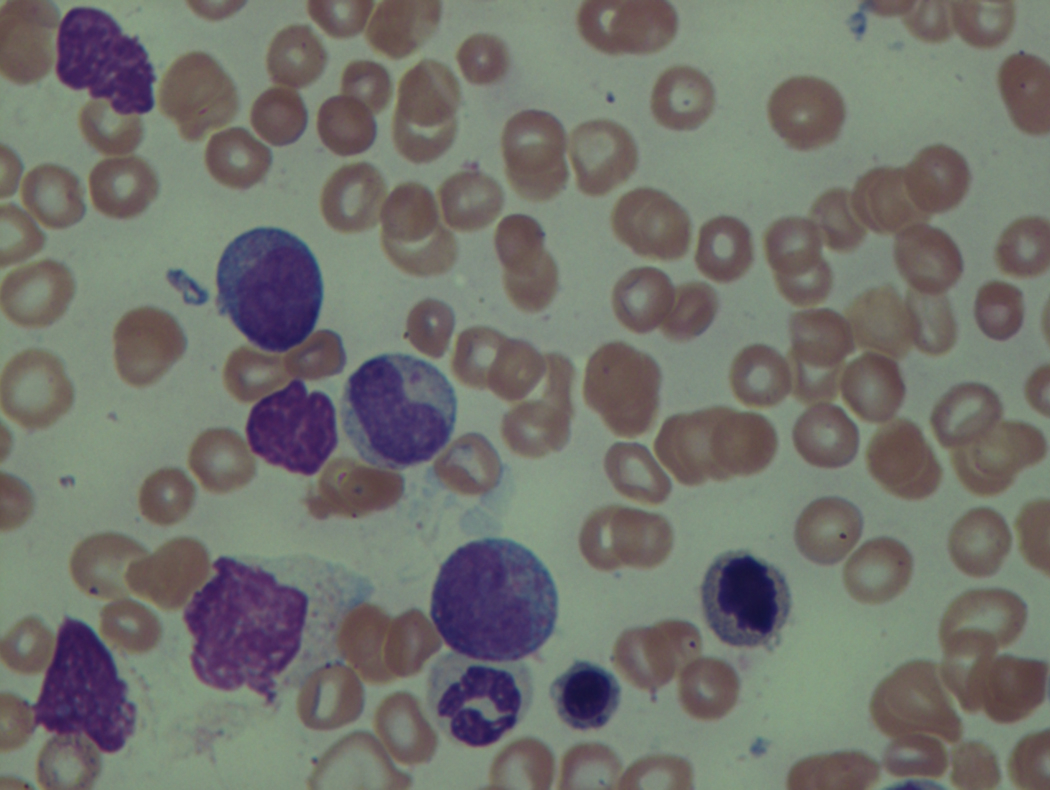
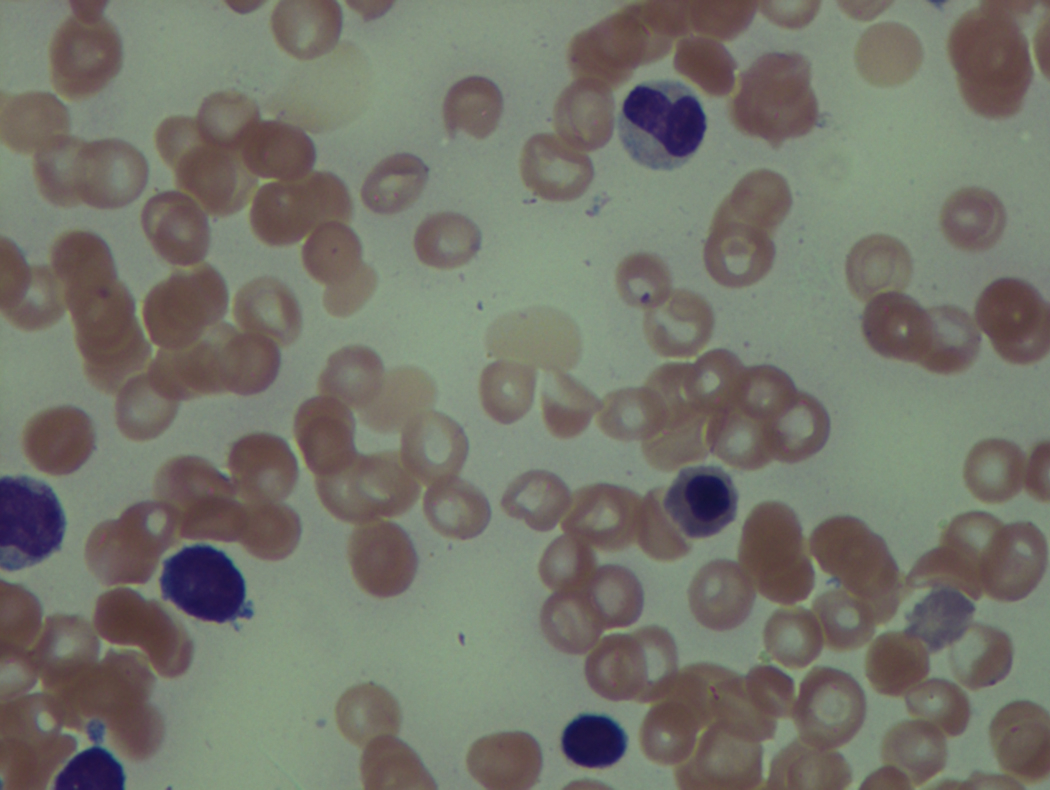
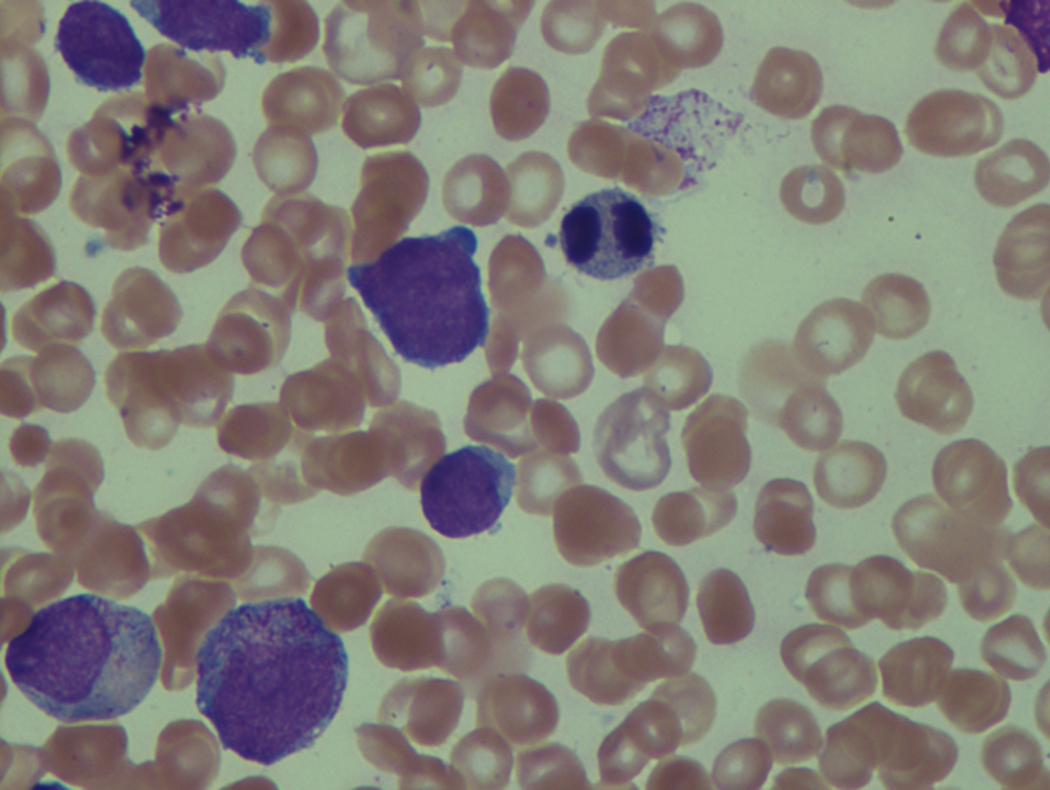
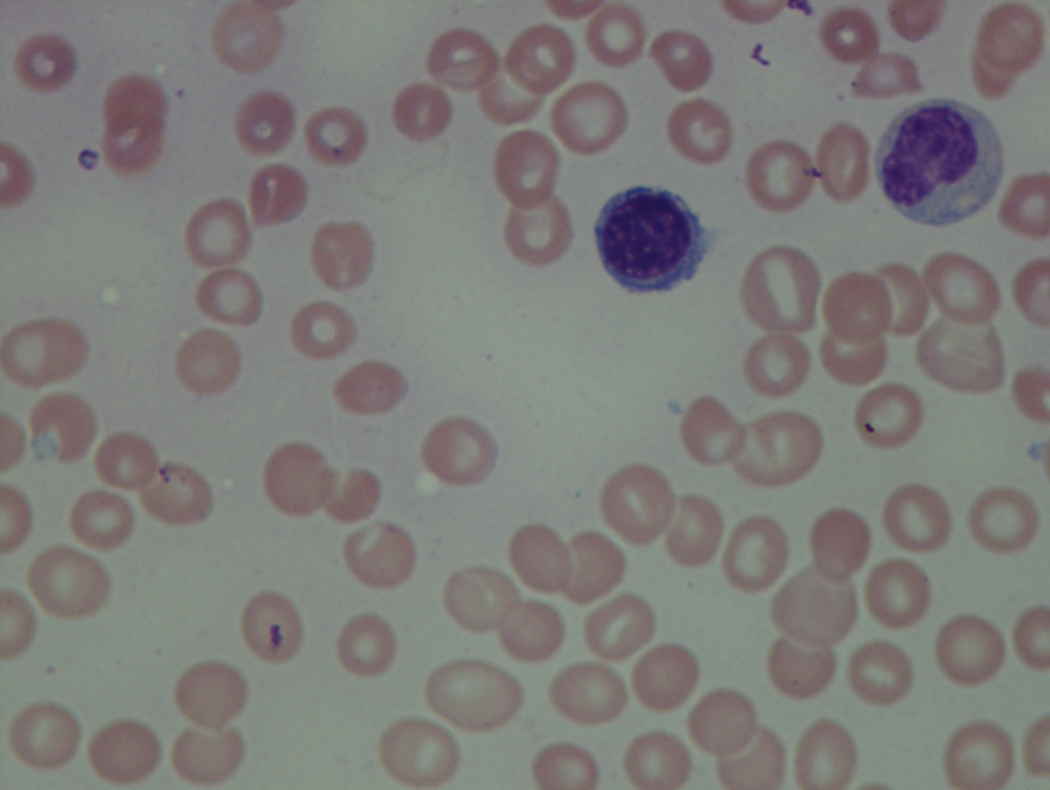
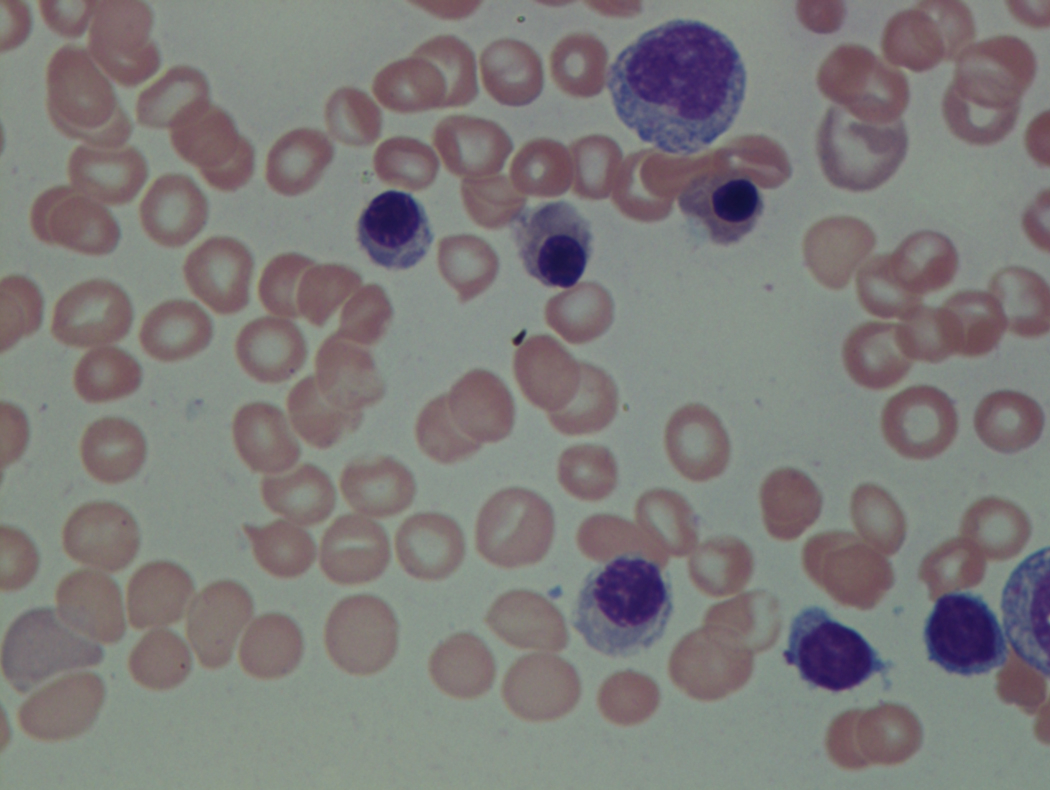
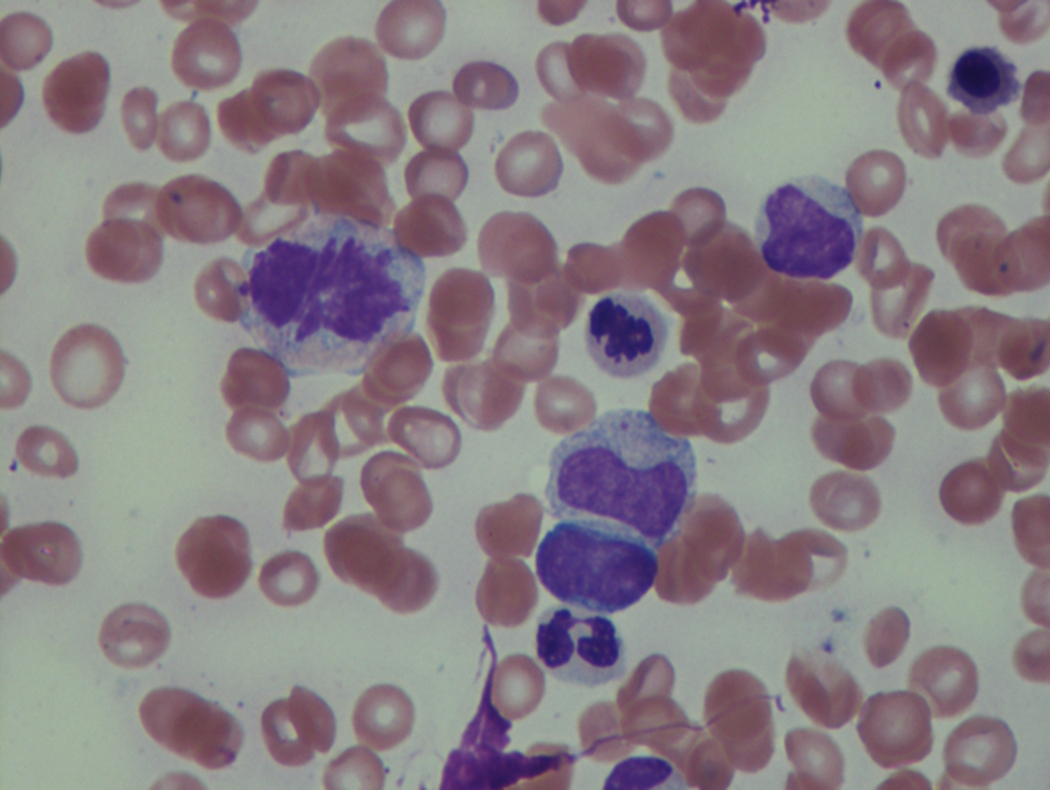

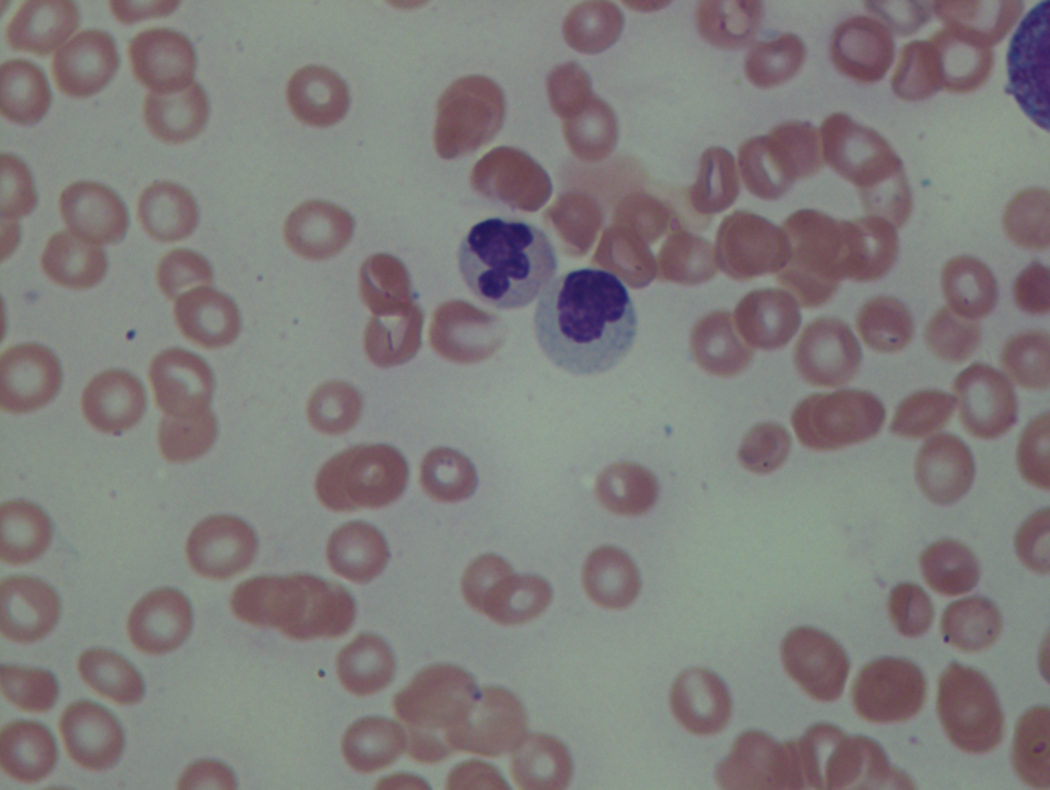
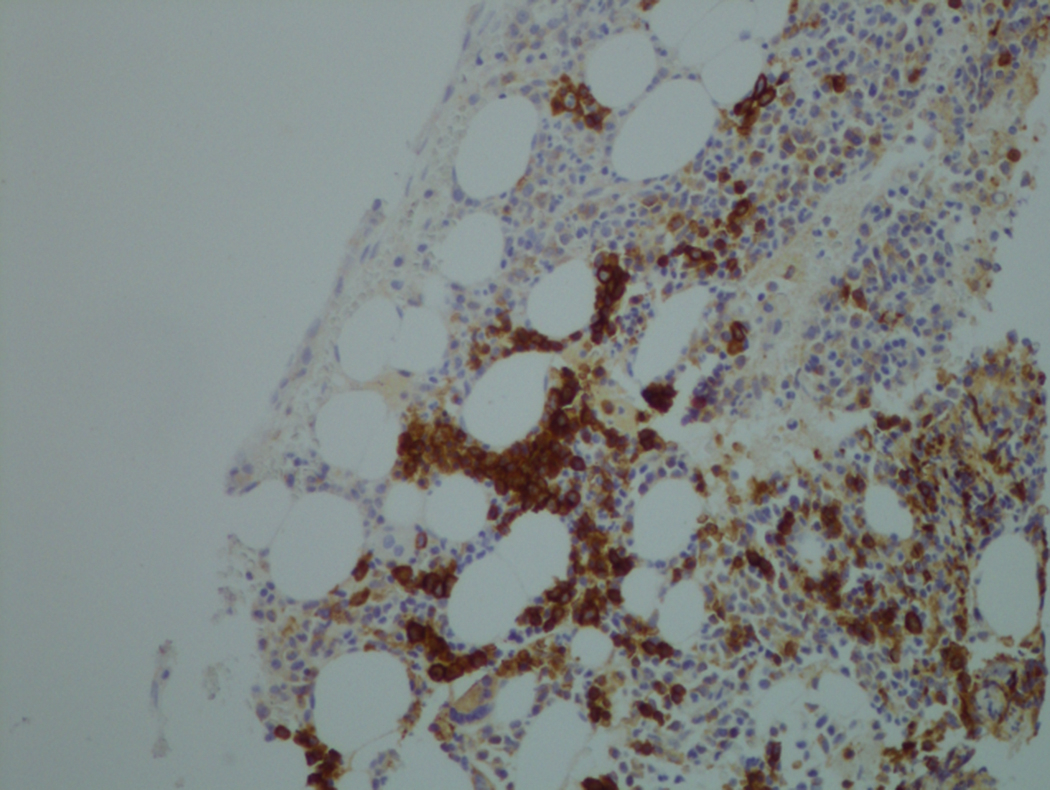
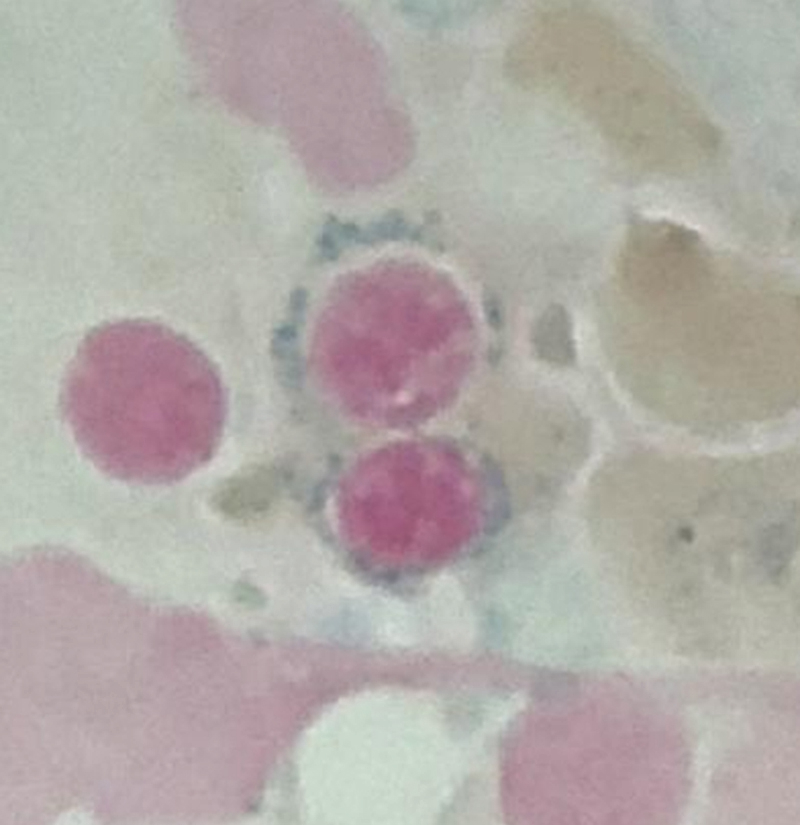
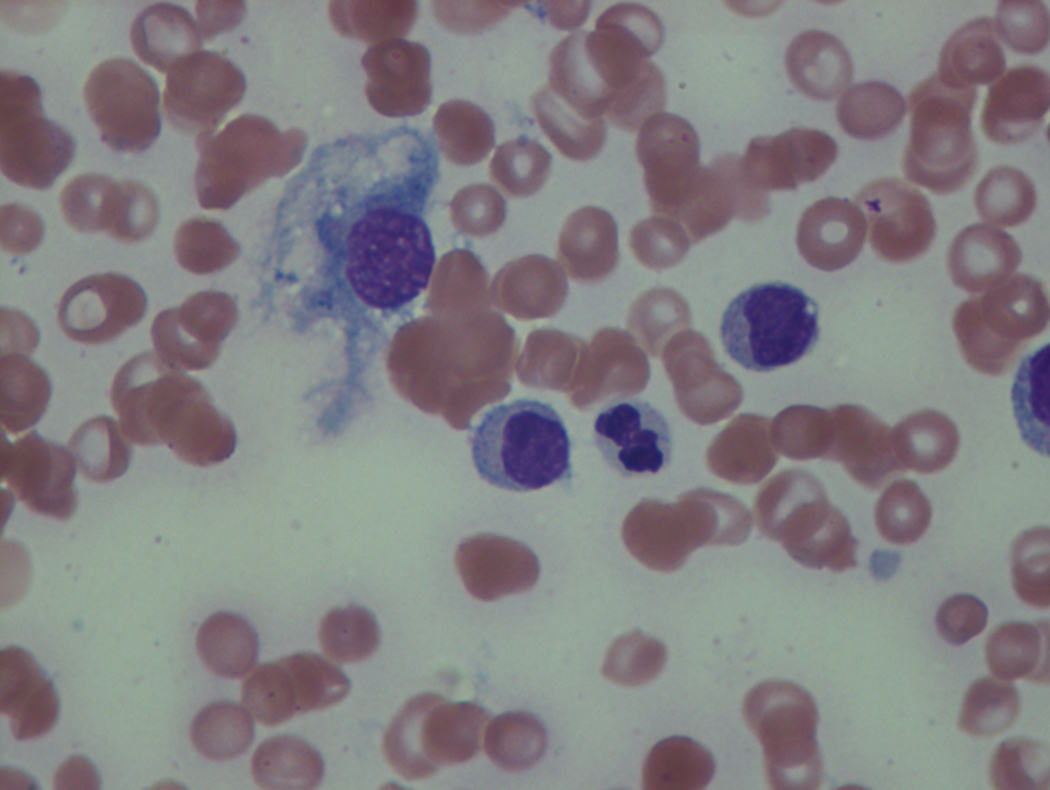
In hypocellular MDS, where all cell lines are decreased, it is important to exclude a diagnosis of aplastic anemia (AA), which may share features of erythroid dysplasia and, less commonly, an abnormal karyotype. A BM biopsy that demonstrates the presence of dysplastic features in megakaryocytes and/or myeloid cells may help differentiate the two. Iron excess is a significant consequence of chronic transfusion requirements in MDS, which may not be present at diagnosis; it is therefore useful to obtain baseline iron studies to exclude iron deficiency. Paroxysmal nocturnal hemoglobinuria (PNH) clones are common in patients with MDS (12.8%–15.5% of MDS patients), but the clinical significance of PNH-type cells in MDS is not well defined. The increased concurrence of these two conditions, along with AA, may be secondary to T-cell mediated suppression of hematopoiesis. Nonetheless, it is important to exclude a concurrent diagnosis of PNH in MDS patients who present with refractory anemia, even in the absence of other clinical indicators such as thrombosis. Flow cytometric analysis of both erythrocytes and granulocytes is the most sensitive and informative assay for diagnosis PNH. Flow is used to document deficiency of glycosyl-phosphatidylinositol-anchored proteins (GPI-APs), most commonly CD55 and CD59 on RBCs and CD11b on monocytes.
Myeloid Cells
Neutropenia is common in patients with MDS, and approximately half of all patients either present or develop absolute neutropenia as a result of their disease progression. Regardless of neutropenic status, patients with MDS are immunocompromised and a major cause of death in this population is infection. The most common infections in MDS are bacterial, usually pneumonia or soft tissue infections. These findings support the concept that even with normal to slightly reduced neutrophil counts, there exists a functional deficit in the immune response in patients with MDS, which tends to worsen and become more clinically significant as the disease progresses.
The presence of monocytosis is important to document in MDS, as it may lead to a diagnosis of chronic myelomonocytic leukemia (CMML) .