Hematopoietic Development
Hematopoiesis is the process by which the various cellular components of the blood are formed. The short-term need for these components can be highly variable, but evidence has accumulated that hematopoietic stem cells (HSCs) are capable of meeting even drastic changes in demand through a variety of differentiation paths.
The development of this capacity is complex. It begins in the embryo with a series of transient hematopoietic waves that occur across several discrete anatomic sites that shift as the embryo evolves . A clear description of how this development occurs is of fundamental importance to our understanding of basic biology and medical science, but our knowledge of embryonic hematopoiesis is still limited compared with what we know of adult HSCs and their microenvironment. This is particularly true for human hematopoiesis, and is reflected in our current difficulties in recapitulating in vitro the development of HSCs from pluripotent stem cells.
In humans, as in other vertebrates, the initial wave of hematopoiesis occurs outside the embryo, in the yolk sac blood islands. These islands are formed by tight groups of mesenchymal cells that originate in the mesoderm but are adjacent to the endoderm; they first differentiate into hematopoietic cells surrounded by endothelial cells in a simple structure that is eventually remodeled to form the branched structure of the yolk sac vascular plexus. The yolk sac primarily produces primitive erythroid cells that express embryonic globins that deliver oxygen to the tissues of the rapidly growing embryo. Large primitive nucleated erythrocytes, with the occasional presence of primitive macrophages and megakaryocytes, represent the major hematopoietic output of the yolk sac from embryonic day 7 to 7.5 in the mouse and 18 days postconception (dpc) in humans. , , This phase of primitive erythropoiesis is transient; it is eventually replaced by adult or definitive hematopoiesis that sustains blood production throughout development and postnatal life.
Hematopoietic activity is first detected within the embryo in a region around the ventral wall of the dorsal aorta called the aorta-gonad mesonephros (AGM). Definitive HSCs, which are marked by serially transplantable activity together with long-term engraftment capacity, emerge alongside non–self-renewing hematopoietic progenitor cells in the AGM region at embryonic day 10.5 in mice and after 5 to 6 weeks of gestation in humans. They are organized in intraaortic hematopoietic clusters (IAHCs) that bud predominantly from the endothelial floor of the dorsal aorta, and accordingly coexpress both endothelial and hematopoietic markers. , As early blood cells develop in close connection with vascular structures (both in the yolk sac and the dorsal aorta), it is believed that a common precursor cell population exists called the hemogenic endothelium (originally referred to as the hemangioblast) that gives rise to both blood and blood vessel cells, which emerge from hemogenic endothelial–expressing cells through an endothelial-to-hematopoietic transition, although definitive proof of this is still awaited. ,
It has yet to be determined whether all hematopoietic stem and progenitor cell (HSPC) subsets and differentiation trajectories are present in the same proportions in each of the embryonic organs, and it is unclear whether HSCs arise in the embryo proper from the AGM or arrive by colonization from the yolk sac. Soon after HSCs emerge, however, hematopoiesis is detected in the fetal liver, followed by the spleen, thymus, and ultimately bone marrow ; it is therefore believed that AGM HSCs (or possibly yolk sac HSCs) first migrate to the liver. The liver rudiment emerges as a diverticulum from the floor of the embryonic gut at early 21 dpc, and from late 22 dpc it contains primitive yolk sac–derived erythrocytes and CD45 + cells, which are likely of monocytic/macrophage lineage. , From 27 to 29 dpc, the liver is seeded by growing numbers of CD34 + /CD45 + cells. In the fetal liver, expansion and differentiation of HSCs enable the development of definitive red blood cells, myeloid cells, and lymphoid cells (including T cells in the thymus and B cells in the marrow), and this organ remains an important niche for hematopoietic differentiation and HSC expansion until birth. Additional hematopoietic activity by HSCs has been found in other tissues, including the placenta, allantois, and umbilical arteries. , , Bone marrow also contains multipotential cells and mesenchymal stem cells (MSCs) that can produce a variety of mesenchymal cell types: osteoblasts (to make bone), chondrocytes (to make cartilage), connective and synovial tissue (to make tendon), and possibly skeletal muscle ( Fig. 1.1 ). There is active research into these and other mesoendodermal bone marrow cell populations, because they suggest that bone marrow could be used to purify and expand these populations for therapeutic benefit in the spleens of mice transplanted with bone marrow cells. ,

Thus the composition and location of the HSPC compartment changes during the developmental stage. Given the importance of environmental signals in eliciting responses to the changing requirements for different blood cell types throughout the growth of the embryo, it seems likely that the activities of HSCs (and their differentiating progeny) are influenced by the dramatic changes in their locations that occur during this period. , Although the role of such extrinsic factors has been difficult to delineate, evidence of intrinsic changes have been easier to infer from comparisons of their behavior when assessed in the same environment either in vitro or in vivo.
Road Maps of Hematopoiesis: Cellular Pathways as Hscs Differentiate Into Terminally Mature Cells
In 1868, Dr. Ernst Haeckel first used the word Stammzelle, or stem cell, to refer to the primordial unicellular organism from which all multicellular life descended. Histopathologists subsequently applied this concept to normal and leukemic hematopoiesis, advancing the hypotheses that red and white blood cells derived from a common progenitor, much as myeloid and lymphoid leukemic cells shared a common precursor. The first in vivo assay for stem cell function was performed in the context of bone marrow transplantation to rescue lethal irradiation. This was followed by the first estimates of HSC population numbers by counting hematopoietic colonies in the spleens of mice transplanted with bone marrow cells. This effort not only provided an estimate of the frequency of spleen colony-forming units at 1 in 10,000 bone marrow cells, but also delivered the first definitive proof of in vivo multipotent progenitor cell function by tracking cytogenetic abnormalities within individual spleen colony-forming units. ,
At the apex of hematopoiesis, a rare population of HSCs sustains all the hematopoietic lineages throughout the lifetime of the individual. Considerable progress has been made over the past three decades in purifying populations of HSCs, the different downstream multipotent progenitors (MPPs), and the unilineage progenitors found in humans and rodents. Fluorescence-activated cell sorting (or FACS) greatly facilitated the purification of transplantable HSCs after a landmark publication from Dr. Irving Weissman’s group in 1988 demonstrated the use of positive and negative selection. Stem cells are self-renewing, and either multi- or unipotent , ; these unique capacities offer opportunities for stem cell–based therapies in the clinic and can be tested via transplantation experiments. Both in mice and humans, HSCs that repopulate in transplantation assays for more than 16 weeks in primary transplantations, and at least as long in the second round of transplantation, are considered to be long-term HSCs (LT-HSCs), whereas progenitors are defined by their lack of extended self-renewal and a restricted lineage differentiation capacity (most are bilineage or unilineage, and so are usually lost within the first 2 to 4 weeks after transplantation). Past research has implied only limited contributions by HSCs to unperturbed hematopoiesis, but HSCs are still believed to be essential to hematopoiesis under recovery or other stress conditions. HSC transplantation has therefore been a key therapeutic strategy in combatting hematologic disorders. As with the stem cells of other tissues, HSCs basically remain quiescent to maintain their undifferentiated state, and their self-renewal capacity is finely tuned by cell intrinsic networks involving key cell cycle regulators and the levels of Hox genes or Polycomb complex protein, along with the activity of transcriptional factors, which together cooperate with cumulative signals from the microenvironment to maintain whole hematopoiesis. , Advances in flow sorting have allowed isolation of relatively pure populations of cells of defined functionality, enabling analysis of the cellular basis of normal hematopoiesis, which has proved critical in advancing our understanding of how the malignant cells of hematologic diseases arise from normal hematopoietic cells through genetic and epigenetic changes.
Human HSCs express the cell surface antigen CD34 (although in mice, HSC capacity has been found to be enriched in CD34 − fractions). CD34 + cells make up approximately 1% of human bone marrow mononuclear cells, and within this group, HSCs principally reside in the CD38 − population, which represents only a small percentage of the CD34 population ( Fig. 1.2 ). Within this CD34 + /CD38 − population, HSCs are enriched in cell populations that both express the cell surface marker CD90 + and do not express CD45RA. This progressive purification scheme allows increasingly greater enrichment of populations of cells exhibiting HSC activity. These highly purified populations can then be tested functionally with in vitro assays, such as the long-term culture-initiating cell (LTC-IC) assay and cobblestone area–forming cell (CAFC) assay, which effectively serve as surrogate assays for stem/early progenitor cells.

For human cells, the most stringent test for HSC capacity is a xenograft study that transplants cell populations into immune-deficient mice (such as Nod Scid gamma mice). Progenitor activity of the transplanted cell populations can be tested in vitro either in colony ( Fig. 1.3 ) or liquid culture assays. Such studies have enabled researchers to construct “road maps” of the variety of cellular intermediates that appear as HSCs differentiate into mature terminally differentiated blood cells , ( Figs. 1.4, 1.5 ). These cellular intermediates exhibit distinctive expression levels of cell surface antigens (immune phenotypes) and, as this remains a very active topic of research, it is likely that the current view of how hematopoiesis proceeds will be modified as more information becomes available. It is also important to bear in mind that some of the information about these cellular pathways comes from studies in rodents and may not apply to human hematopoiesis.


Around the year 2000, after critical advances in the characterization of progenitor populations downstream of HSCs, a consensus model emerged of the hematopoietic differentiation tree (see Fig. 1.4 , left panel). , , , In this model, the first branch point segregates lymphoid potential from all other lineages (myeloid, erythroid, and megakaryocytic), followed by further branching steps on either side of the tree progressing from multipotent to bipotent and finally to unipotent progenitor cells. In this model, HSC pools are divided into LT-HSCs (which provide long-term engraftment and will serially engraft irradiated mice) and short-term HSCs (ST-HSCs) that have more limited self-renewal capacity in a serial transplant assay. But beyond this basic classification, the pathways of differentiation remain under debate. HSCs are believed to give rise to an MPP capable of producing all blood cell lineages but lacking the capacity for serial transplantation in mice. This MPP then differentiates into the myeloid and lymphoid lineages. The common myeloid progenitor (CMP) gives rise to all myeloid cells. Similarly, the common lymphoid progenitor (CLP) can differentiate into B and T lymphocytes and natural killer (NK) cells. The CMP further differentiates into two progenitors with more restricted differentiation potential: the megakaryocyte-erythroid progenitor (MkEP) and a granulocyte-macrophage progenitor (GMP).
The subsequent identification of other surface markers has suggested several modifications of this classical tree. Lymphoid and myeloid fates now remain linked prior to separation further down the tree, whereas megakaryocyte branching arrives earlier, along with subdivision of the MPP compartment into distinct subpopulations. Other aspects of the model are being reconsidered; the first lineage commitment step from the HSC pool, for instance, may not be a lymphoid-myeloid differentiation decision step but rather one that allows the megakaryocyte-erythroid lineage to split off, leaving a progenitor with both lymphoid and myeloid (granulocyte-macrophage) potential. Termed a lymphoid-primed MPP (LMPP), this cell then differentiates into the granulocyte-macrophage and lymphoid lineages. T-cell/myeloid progenitors and B-cell/myeloid progenitors have now been isolated, and mounting evidence indicates that cell intermediates with combined myeloid/lymphoid potential may exist. Moreover, the existence of mixed myeloid/lymphoid leukemia supports the notion that there may be a normal counterpart of this malignant population. Both models are believed to operate as shown in the middle panel in Fig. 1.4 .
Moreover, increasing evidence suggests that the picture may be even more complicated, as the HSC pool itself is functionally and molecularly heterogeneous. HSCs have been identified retrospectively after single-cell transplantation by clonal assays, and these assays have demonstrated the heterogeneity of currently available HSC-enriched fractions. Using limiting dilution analysis and single-cell transplantation, the Dykstra et al. research team has described HSCs that differ in their relative myeloid and lymphoid output. More recently, various research groups identified platelet-biased HSCs that predominantly differentiate toward megakaryocytes and platelets, and subsequent transplantation experiments revealed that single HSCs execute only a limited repertoire of lineage fate patterns, with platelets providing the only observed long-term unilineage read-out. Yamamoto’s group further proposed an additional differentiation model in which HSCs can directly differentiate into lineage-restricted progenitors while bypassing the MPP stage during acute conditions that demand rapid replenishment of mature cells (as in response to ablation stress). , Notably, phenotypic HSCs comprise a major source of the megakaryocyte/platelet lineage in steady-state conditions, but these cells show multilineage differentiation capacity once they are transplanted into irradiated recipient mice. These data imply potential differences in fate decision mechanisms between steady state and hematopoietic recovery. Indeed, theories differ regarding the contributions of HSCs to unperturbed homeostasis versus tissue recovery conditions, and technical considerations may influence conclusions derived from transplantation experiments.
In vivo transplantation assays, which are the only assays that can test for HSC self-renewal, have proved fundamental to recent advances in our knowledge of HSC biology. However, because the mouse microenvironment cannot optimally support human cells, xenotransplants do not robustly read out all possible differentiation routes, particularly at the single-cell level. The past 10 years have therefore seen a collective effort to define the lineage potential of single human progenitor cells by using highly defined in vitro models that can support the differentiation of most mature blood cell types. Work in other laboratories suggested that the first restriction in lineage potential does not segregate lymphoid and myeloid potential, as postulated by the common myeloid and lymphoid progenitor models, but rather allows these potentials to remain coupled in the lymphoid-primed MPP and myeloid-lymphoid progenitor compartments. Many other subpopulations, most with either bilineage or unilineage capacity, have since been found in both the lympho-myeloid branch and the myelo-erythroid-megakaryocytic branch. Taken together, these findings indicate that few single cells read out as multipotential within the progenitor compartment. There are, however, subtle issues in such interpretations, because even though a given cell may appear to be multipotent based on its molecular state, if it makes a lineage choice before dividing, it will be defined as unipotent in functional assays. Nevertheless, growing evidence implies that lineage choice occurs earlier than previously thought, and as recently shown for dendritic cells, probably as early within phenotypic HSC populations.
Although it is likely that all these structures capture true aspects of HSC differentiation, collectively they would be difficult to squeeze into a single, rigidly branching tree. New ways of graphically representing the process of HSC differentiation are thus required. Recent advances in single-cell transcriptomic snapshots have suggested a new model of differentiation in which acquisition of lineage-specific fates is a continuous process, and unilineage-restricted cells emerge directly from a continuum of low-primed, undifferentiated HSPCs, without major transitions through the multipotent and bipotent stages , , ( Fig. 1.4 , right panel). It is also clear that genetic changes (such as mutations) or epigenetic changes, either from disease states or experimentally induced, can alter the progress of cells through these compartments. There are, however, caveats regarding this new continuum model and its heavy reliance on single-cell RNA-sequencing (scRNA-seq), as cell surface markers can highly enrich for particular behaviors (differentiation, self-renewal, or proliferative output). The biological validation of a model in which all lineages branch out directly from the HSC compartment would require additional supportive evidence.
Once specified, HSCs have the option of progressing down a number of cell fate paths. Two key features of HSCs are their ability to self-renew throughout the lifetime of the organism and their ability to maintain their multipotentiality. Most of the time, HSCs remain quiescent and are in phase G 0 of the cell cycle, although they continue to be capable of undergoing cell divisions as required. HSC divisions result in either self-renewal or differentiation, with the balance between the two directly impacting hematopoietic homeostasis. , , Their possible division options are (1) symmetric self-renewal expansion (symmetric division [SD]; both daughter cells have the same function as the original cell), (2) self-renewal maintenance (asymmetric division [AD]), and (3) differentiation (symmetric commitment [SC]; both daughter cells are differentiated from the original parent cell). These cell fate decisions can be evaluated by paired daughter cell assays, and the eventual division pattern of a population of HSCs is determined by the in vivo repopulation capacity of its daughter cells. , , , A fourth option is that an HSC can undergo apoptosis. The sum of the cell fate decisions of all an organism’s HSCs determines the quality and quantity of its hematopoietic activity.
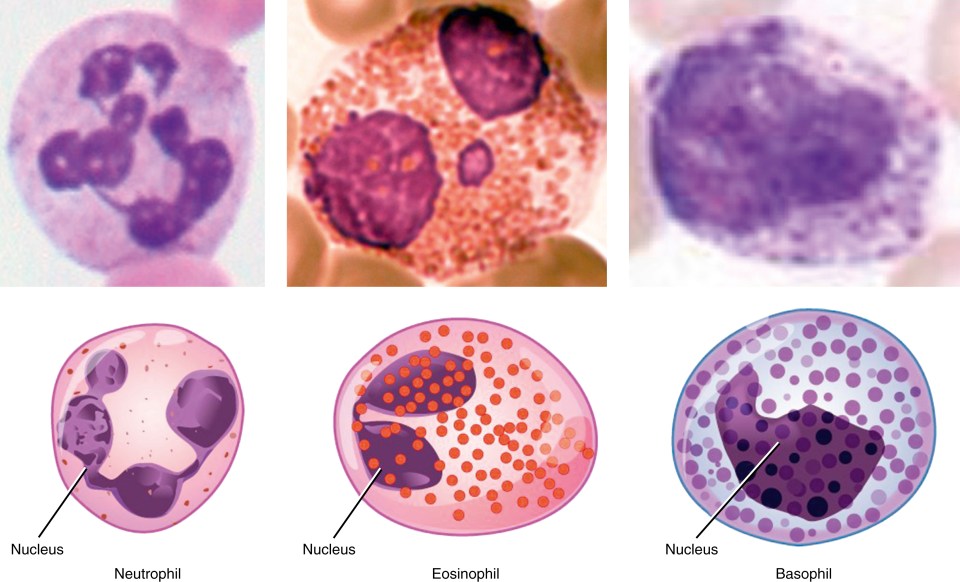
As HSCs differentiate into progenitors and then terminal mature cells, changes occur in the cell fate options that cells can make ( Figs. 1.4, 1.5 ). For example, progenitors have reduced self-renewal potential and this further declines with differentiation. In contrast to HSCs, progenitors spend much less time in phase G 0 and are highly proliferative. They provide much of the amplification in cell numbers required to satisfy the enormous daily demand for blood cells. In an adult human, approximately 10 10 new blood cells are made daily. Finally, as progenitors enter terminal differentiation, their proliferative capacity is slowly lost, and cells enter progressively more restricted pathways of differentiation that eventually lead to unilineage differentiation. Terminally mature cells are often postmitotic (e.g., red cells and granulocytes) and express only the proteins required for function by terminally mature cells.
Transcriptional Control of Hematopoiesis
This increasingly narrow lineage potential results from a precise combination of gene expression signatures and epigenetic modification. Proteins known as transcription factors (TFs), which regulate the transcription of genetic information and are expressed either exclusively in blood cells or in restricted tissue-specific patterns, are critical to the control of hematopoiesis. , The crucial role of these TFs has been recently highlighted by the discovery that in hematologic diseases, such as lymphoma and leukemia, the genes encoding these TFs exhibit acquired mutations. The importance of these TFs is also underscored by the conservative role they have played in hematopoiesis throughout evolution. Because purifying and biochemically interrogating very rare HSC populations has proved so difficult, the roles of specific TFs in HSC fate decisions have instead been investigated in animal models in which genes encoding critical TFs have been deleted, modified, overexpressed, or misexpressed ( Fig. 1.6 ). The key points arising from these studies have been as follows:
- 1.
TFs are divided into families that have similar protein domains.
- 2.
TFs often bind DNA and interact with other proteins (other TFs and proteins that control transcription) via specific domains.
- 3.
TFs work in combination to both activate and repress the expression of a large number of genes.
- 4.
TFs are required at discrete stages of hematopoiesis, and any one TF can function at multiple stages within one lineage and can also function in more than one lineage.
- 5.
Ultimately, TFs work in complicated networks that can be modeled much like semiconductor/computing networks. TFs work in negative feedback loops, feed-forward loops, and cross-antagonistic loops, to mention just three such types of interaction.
- 6.
The function of TFs helps regulate an HSC’s potential to make blood cells of different lineages, proliferate, undergo apoptosis, and self-renew.





