INTRODUCTION
Plasma cell dyscrasias are heterogeneous disorders arising from the proliferation of a monoclonal population of plasma cells. Some of these disorders can present serendipitously as benign processes that can be observed; others are highly aggressive and require immediate intervention. The most common plasma cell dyscrasia is monoclonal gammopathy of undetermined significance (MGUS), a benign condition that can be observed. Related disorders include smoldering multiple myeloma (SMM), multiple myeloma (MM), solitary plasmacytoma of the bone, extramedullary plasmacytoma, Waldenström macroglobulinemia (WM), primary amyloid light-chain (AL) amyloidosis, heavy-chain disease, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes) syndrome, and the recently recognized TEMPI (telangiectasias, elevated erythropoietin and erythrocytosis, monoclonal gammopathy, perinephric fluid collection, and intrapulmonary shunting) syndrome. The spectrum of MGUS, SMM, and MM represents a natural progression of the same disease. This chapter focuses on the etiology, genetics, biology, diagnosis, clinical features, and current therapy of MM and other plasma cell disorders.
Major recent discoveries have changed the way we understand, diagnose, and treat plasma cell dyscrasias. The initial sequencing of the myeloma genome and single-cell genetic analysis paved the way for the concept of intraclonal heterogeneity and Darwinian selection of clones. Increasingly sensitive diagnostic and monitoring techniques allow for more accurate diagnosis, minimal residual disease monitoring, and detection of early relapse. New diagnostic criteria for MM have been implemented, and the introduction of novel classes of agents such as immunomodulatory drugs and proteasome inhibitors has led to improved overall survival. Additionally, immunotherapy using monoclonal antibodies against different myeloma targets has shown promising activity in clinical trials. Major advances have also occurred in WM as a highly recurrent single point mutation of the MYD88 gene has been identified, and new treatments that abrogate this highly active pathway are already in use. Finally, a new paraneoplastic syndrome, the TEMPI syndrome, has been identified and described.
MULTIPLE MYELOMA
Multiple myeloma is a malignant proliferation of plasma cells. In virtually all cases, myeloma cells (as well as their precursors MGUS and SMM) secrete immunoglobulins. Usually, myeloma cells secrete immunoglobulin (Ig) G (60%); other types are less common (IgA 20%, IgD 2%, IgE <0.1%, biclonal <1%). Light chain–only secretion is noted in 18%; <5% of patients do not secrete a heavy- or light-chain immunoglobulin (nonsecretory MM).
In 2014, approximately 24,000 people were diagnosed with MM in the United States, and 11,090 died from the disease. The median age at diagnosis is 69 years. The incidence is highest in the age range of 65 to 74 years (27.7%), followed by the 75- to 84 year-old range (24.7%). The annual age-adjusted incidence of the disease per 100,000 population is 7.2 among white men and 4.3 among white women. Among African Americans, the frequency doubles to 14.8 in men and 10.5 in women. There is also a difference in mortality by racial group. The annual age-adjusted mortality rate per 100,000 is 4.0 and 2.5 in white men and women, respectively, and 7.7 and 5.3 in African American men and women, respectively. The incidence and mortality rates are lowest among Asians and Pacific Islanders.
Risk factors that predispose to MGUS and MM point toward common shared etiologic environmental and genetic factors. Age is a risk factor for MGUS, because its prevalence is four times higher among individuals ≥80 years old than among those 50 to 59 years old. Increased risk of MGUS has also been reported in first-degree family members of patients with MGUS and MM (risk ratio between 2 and 3). In a study of black and white women of similar socioeconomic status, obesity, black race, and increasing age conferred an increased risk of MGUS. Personal and family history of autoimmune or inflammatory disorders as well as infections have been linked to an increased risk of MGUS and MM. Exposure to infections has been hypothesized to be involved in the malignant transformation of MM, or it could represent impaired immunity associated with MGUS and SMM, which often precedes a diagnosis of MM. Radiation exposure, pesticides, and cleaners are also associated with an increased risk of MGUS and MM.
Although MM is not an inherited disease, more than a hundred familial cases have been reported in the literature. The largest series described 39 unique families with 79 MM cases. Both dominant and recessive inherited traits may play a role in familial MM. Large genomic studies have identified low penetrant genetic variants that confer a modest increase in the risk of developing MM (1,2). Based on epidemiologic and familial aggregation studies, most of the inherited risk of developing MM may result from different genetic polymorphisms, each of which has only a small effect on the predisposition to develop disease (3).
Multiple myeloma arises from terminally differentiated B cells or even early committed B cells (germinal center B cells) that manifest clinically as more differentiated plasma cells. The major role of normal differentiated plasma cells is to produce immunoglobulins (antibodies) to fight infections. To become an effective part of the adaptive immune system, B cells must undergo immunoglobulin gene rearrangement and affinity maturation in response to antigens presented by antigen-presenting cells within the lymph node germinal center. For this to occur, hypervariable regions in the immunoglobulin heavy chain locus (IGH in chromosome 14q32) undergo programmed mutations (somatic hypermutation) through which, among others, double DNA strand breaks and chromosomal translocations are generated. The primary etiology of MM has been linked to IGH translocations and increased copies of odd-numbered chromosomes (hyperdiploidy), which result in cyclin D dysregulation. These events can be observed early in the course of monoclonal gammopathies (such as in MGUS or SMM) as well as in MM, suggesting that they are primary genetic events. Initial whole-genome and exome sequencing in 38 MM patients confirmed the complexity of genetic alterations seen in MM and uncovered secondary mechanisms of transformation to MM (4). Secondary events included mutations in the oncogene MYC (most commonly observed in plasma cell leukemia or aggressive forms of MM), mutations in the nuclear factor-κβ (NF-κβ) pathway, including BRAF and RAS, and chromosome copy number abnormalities such as deletions, amplifications, or additions. Changes in DNA methylation patterns are also important secondary events leading to increased tumor diversity and more aggressive forms of plasma cell dyscrasias (Table 11-1).
Primary genetic events IGH translocations [t(4:14), t(6:14), t(14:20), t(14;16), t(11,14)] Hyperdiploidy (trisomies of chromosomes 3, 5, 7, 9, 11, 15, 21) |
Secondary genetic events Additions (1q, 17q, 12p) Deletions (1p, 13, 11q, 14q, 17p, 6q, 8p) Translocations [t(8;14)] Methylation changes (global hypomethylation and gene-specific hypermethylation) Mutations in NF-κβ pathway (TRAF3, I-κβ) Proliferation (NRAS, KRAS, BRAF, MYC, MAPK, PI3K, MET) |
Different tests for gene expression profiling (GEP) are available for molecular classification of MM. Currently, molecular profiling of MM is mostly used for research purposes (eg, identification of high-risk MM for inclusion in clinical trials). These tests may become increasingly important as we develop more personalized treatment for MM.
Serial genomic analysis during the disease course of myeloma patients has identified different MM subclones within the same tumor. This has been termed intraclonal heterogeneity. In this model, different myeloma subclones compete for selection as they are exposed to the microenvironment and therapeutic pressures (5). Single-cell genetic analysis at diagnosis confirmed that MM is highly heterogeneous and characterized by the accumulation of a diverse range of mutations at the subclonal level (6). In this scenario, the acquisition of new mutations leads to new subclones with different clinical phenotypes and sensitivities to therapy. Intraclonal heterogeneity in myeloma has many potential implications for therapy, suggesting that subclonal targeting in combination therapies may be needed to eradicate the multiple subclones. Increasing genetic complexity is seen with progression from MGUS to MM and plasma cell leukemia, which may suggest that earlier treatment may result in improved clinical outcomes.
The bone marrow microenvironment also plays a role in the etiology of MM and its related disorders. Plasma cells communicate effectively with the microenvironment in a process called cell trafficking. Upregulation of cytokines that increase vascular permeability, proliferation, or cell homing (interleukin [IL]-6, vascular endothelial growth factor, and insulin-like growth factor) have been involved in the progression to MM. Gene expression profiling has revealed that modulation of certain genes can lead to a permissive microenvironment that promotes growth of myeloma subclones leading to active disease (7). Thus, targeting the microenvironment is an area of extensive research that, combined with therapeutic targeting of myeloma subclones, may lead to improved outcomes. New and effective antimyeloma combination therapies and well-designed clinical trials are needed to test these hypotheses.
The clinical presentation of MM and its precursors is variable. Patients with MGUS or SMM usually do not present with specific myeloma-related symptoms. Their diagnosis is often incidental based on workup for a low albumin-to-globulin ratio, high serum protein, or other conditions such as autoimmune diseases, peripheral neuropathy, skin rashes, or hemolytic anemias.
In contrast, patients initially presenting with MM usually have at least one of the CRAB criteria (hyperCalcemia, Renal disease, Anemia, and Bone disease) classically used to define symptomatic MM. Anemia is the most common finding, occurring in 73% of patients, and is typically a normocytic, normochromic anemia. Anemia can be due to a variety of factors, including marrow replacement or cytokine production by plasma cells, which lead to decreased erythropoiesis, or decreased erythropoietin levels due to renal disease (8).
Bone pain is common, occurring in 60% of patients, and related to increased resorption of bone, leading to lytic bone lesions. Painful vertebral compression fractures can occur and may represent a medical emergency when associated with symptoms of cord compression. Increased bone resorption has been attributed to factors such as RANK ligand (RANKL), osteoprotegerin (OPG), macrophage inflammatory protein (MIP)-1α, IL-6, and IL-3, which stimulate osteoclast activity in areas infiltrated by plasma cells as a result of interactions between plasma cells and the microenvironment (Fig. 11-1).
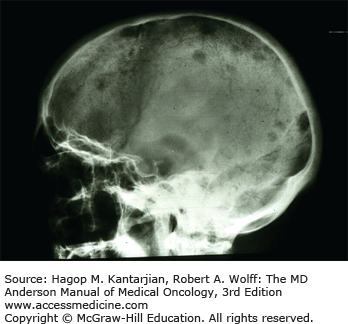
An elevated creatinine is a presenting sign in 50% of patients. Renal disease is often attributed to light-chain cast nephropathy resulting from precipitation of light chains that bind to Tamm-Horsfall mucoproteins secreted by cells in the ascending loop of Henle. These precipitated complexes obstruct the distal convoluted tubules and collecting ducts, leading to tubular atrophy and interstitial fibrosis. Other causes of renal failure include hypercalcemia, leading to nephrocalcinosis, as well as amyloidosis, heavy-chain disease, and light-chain disease.
Hypercalcemia >11 mg/dL is present in 10% of patients and represents a medical emergency requiring hydration with isotonic saline and bisphosphonate therapy with zoledronic acid or pamidronate in moderate or severe cases. Calcitonin can also be used to rapidly reduce serum calcium levels.
Other common presenting symptoms include fatigue (32%) and weight loss (20%). Due to immune dysfunction, patients are at risk for infections. About 7% to 18% of patients may present with extramedullary plasmacytomas. Less common symptoms include fever, splenomegaly, hepatomegaly, and lymphadenopathy.
Once a plasma cell dyscrasia is suspected, a comprehensive diagnostic workup should be initiated to demonstrate the presence or absence of a clonal plasma cell disorder, to determine if end-organ damage is present, and to evaluate laboratory markers related to prognosis. These should include the following components.
Complete blood count (CBC)
Serum chemistries including creatinine, calcium, albumin, lactate dehydrogenase (LDH), β2-microglobulin, and immunoglobulin levels (IgG, IgA, IgM)
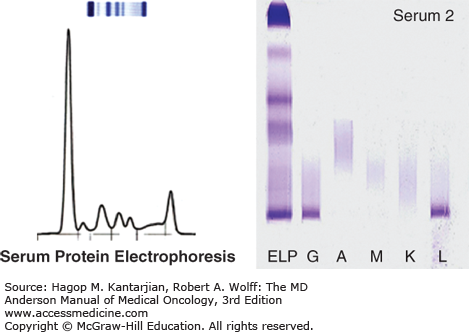
Serum protein electrophoresis with immunofixation to quantify monoclonal protein (M-protein) and determine immunoglobulin isotype
Serum free light-chain assay to evaluate the ratio of serum kappa to lambda light chains
Urinalysis with 24-hour urine collection with protein electrophoresis and immunofixation (Fig. 11-2)
Skeletal survey with plain films of the axial and appendicular skeleton is the minimum standard of care to evaluate lytic bone lesions.
Advanced imaging with either whole-body low-dose computed tomography (CT), positron emission tomography–computed tomography (PET-CT), or magnetic resonance imaging (MRI) can detect up to 80% more lesions compared with plain film x-rays.
– An advanced imaging modality is particularly recommended in the diagnosis of SMM to detect subtle bone lesions that would warrant the initiation of treatment. It is also helpful in assessing baseline disease burden as an adjunct to serum and urine markers prior to initiation of treatment in MM.
– A CT scan can be helpful in the characterization of soft tissue masses in the case of extramedullary plasmacytomas and can direct to an area to be biopsied.
– An MRI scan is useful for evaluating the axial skeleton in the presence of symptoms and assessing for spinal cord compression. It can also identify abnormal marrow uptake as T1-weighted images will show a diffuse decrease in marrow signal intensity but will enhance with the administration of contrast.
Positron emission tomography–computed tomography can be prone to false-positive findings but has more specificity due to increased metabolic uptake at the site of lytic lesions and is the preferred initial baseline advanced imaging modality at the University of Texas MD Anderson Cancer Center (MDACC) in combination with skeletal surveys.
There is no role for nuclear bone imaging because bone scan isotopes are not taken up by lytic lesions.
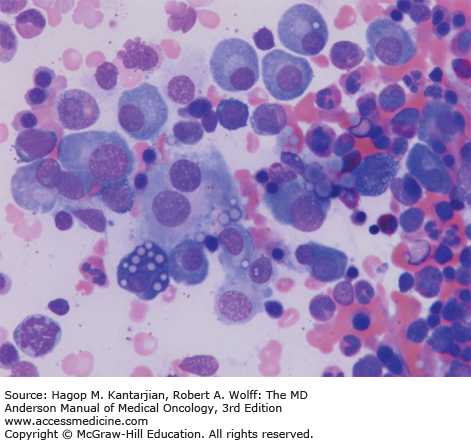
Morphologic review and immunohistochemistry (Fig. 11-3)
Flow cytometry for immunophenotyping of plasma cells:
– Plasma cells are positive for CD38 and CD138.
– Normal plasma cells have higher expression of CD19 and CD45; malignant plasma cells typically lack these surface antigens.
– Malignant plasma cells have increased expression of CD56 and CD117; normal plasma cells have weak expression of these markers.
Conventional cytogenetic karyotyping
Fluorescent in situ hybridization (FISH) for recurrent chromosomal deletions, amplifications, and translocations that have prognostic significance; these include:
– Deletion 13q14, deletion 17p13 (TP53), and deletion of 1p32
– Amplification of 1q21
– Translocations involving the immunoglobulin heavy-chain locus on chromosome 14q32 and its common partners, including 11q13 (CCND1), 4p16 (FGFR3 and MMSET), 16q23 (c-MAF), 6p21 (CCND3), and 20q12 (MAFB)
Gene expression profiling of the CD138+ bone marrow aspirate plasma cells to identify high-risk MM and to facilitate inclusion in clinical trials
Abdominal wall fat pad biopsy (warranted if there are signs and symptoms suggestive of amyloidosis; see separate discussion), which should be stained with Congo red stain. Amyloid fibrils show green birefringence under polarized light.
Serum viscosity (if there are concerns for hyperviscosity usually due to elevated IgM levels in WM; see separate discussion). Hyperviscosity should be a clinical diagnosis, and therapeutic plasma exchange should not be delayed while waiting for the results of serum viscosity level.
Based on the above workup, a diagnosis of a plasma cell dyscrasia may be made, which can span the spectrum of the premalignant MGUS stage to SMM to full malignant transformation to MM. Definitions of these clinical stages according to the International Myeloma Working Group (IMWG) criteria are summarized in Table 11-2 (9). Historically, SMM and MM have been distinguished by the presence of end-organ damage as defined by CRAB criteria. The 2014 updated IMWG criteria were revised to reclassify some SMM patients as having MM (even in the absence of symptoms) if certain biomarkers were present that might indicate impending development of CRAB features. These include clonal bone marrow plasmacytosis ≥60%, an involved-to-uninvolved serum free light chain ratio ≥100, or more than one focal lesion on MRI studies of at least 5 mm in size. Patients with SMM and at least one of these biomarkers have a 70% to 80% chance of progression to MM at 2 years compared to 20% (10% per year) in the absence of these high-risk features. Initiating therapy in these patients may delay the onset of MM-defining end-organ damage events and associated morbidity and adverse effects on quality of life.
| Definition | Progression Rate | ||
|---|---|---|---|
| Premalignant | Monoclonal gammopathy of undetermined significance (MGUS) |
|
|
| Smoldering multiple myeloma (SMM) |
| 10% per year (see Table 11-9 for risk stratification in SMM) | |
| Multiple myeloma (MM) |
| Not applicable |
The course of MM is heterogeneous. Risk stratification using staging and prognostic tools may yield insights into the underlying disease biology and expected course. Prognostic studies can help stratify patients in clinical trials and may help guide therapy [eg, bortezomib in t(4;14) and del 13q].
The International Staging System (ISS) was established in 2005 by the IMWG after a retrospective analysis of the outcomes of >10,000 patients across 17 different centers. In this study, β2-microglobulin and albumin were powerful correlates of median survival, and patients could be categorized into three stages based on serum levels at diagnosis (Table 11-3). Because β2-microglobulin is renally excreted, high levels may be found in the presence of renal failure, which makes the interpretation of the ISS in this setting challenging. The ISS is the current preferred staging method and has supplanted the previously used Durie-Salmon staging system, which was confounded by observer-dependent variables, such as degree of lytic bone lesions, that added subjectivity in its assessment. It is important to note that the ISS has only been validated at the time of diagnosis in patients with MM and should not be extrapolated to patients with MGUS or SMM.
|
|
|
In addition to the ISS, patients can be stratified into standard-, intermediate-, and high-risk categories based on cytogenetic findings by both conventional karyotyping and FISH. Other high-risk features include elevated serum LDH, extramedullary disease, circulating plasma cells, and a high-risk GEP pattern as defined by a 70-gene panel. Risk stratification based on these criteria is summarized in Table 11-4.
| Standard Risk | Intermediate Risk | High Risk |
|---|---|---|
t(11;14) t(6;14) Hyperdiploid karyotype | t(4;14) Del 13q Hypodiploid karyotype | Del 17p13 Amplification of 1q21 t(14;20) t(14;16) Lactate dehydrogenase ≥2× institutional upper limit of normal Plasma cell leukemia High-risk gene expression profiling signature |
The IMWG proposed new guidelines in 2006 to standardize response criteria in MM and to define disease progression to facilitate comparisons of outcomes between treatment centers and for reporting results in clinical trials. These International Uniform Response Criteria guidelines are summarized in Table 11-5. Assessment of response with M-protein measurements using serum protein electrophoresis, urine protein electrophoresis, and serum free light-chain assay is recommended prior to each cycle of therapy. Bone marrow biopsy is necessary to monitor disease in the absence of a measurable M-protein in the serum or urine or to document a complete or stringent complete response. Serial imaging assessments may be required if soft tissue plasmacytomas are present at baseline.
| Response Category | Criteria |
|---|---|
| sCR | Meets criteria for CR PLUS Normal FLC ratio AND No clonal cells in bone marrow by immunohistochemistry or immunofluorescence |
| CR | Negative serum and urine immunofixation AND Disappearance of any soft tissue plasmacytomas AND ≤5% plasma cells in bone marrow |
| VGPR | Serum and urine M-protein detectable by immunofixation but negative M-protein OR ≥90% reduction in serum M-protein plus urine M-protein level <100 mg per 24 h |
| PR | ≥50% reduction of serum M-protein and reduction in 24-h urinary M-protein by ≥90% or to <200 mg per 24 h If unmeasurable serum and urine M-protein, ≥50% decrease in the difference between involved and uninvolved FLC levels If unmeasurable serum and urine M-protein serum FLC assay, ≥50% reduction in plasma cells is required in place of M-protein, as long as baseline bone marrow plasma cell percentage was ≥30% In addition to above criteria, a ≥50% reduction in the size of any baseline soft tissue plasmacytoma is required |
| SD | Not meeting criteria for CR, VGPR, PR, or progressive disease |
| PD | Increase of ≥25% from baseline in at least one of the following: Serum M-component (the absolute increase must be ≥0.5 g/dL) Urine M-component (the absolute increase must be ≥200 mg/24 h Difference between involved and uninvolved FLC levels if serum and urine M-protein are unmeasurable (the absolute increase must be >10 mg/dL) Bone marrow plasma cell percentage (the absolute % must be ≥10%) Development of new bone lesions or soft tissue plasmacytomas or definite increase in size of existing bone lesions or soft tissue plasmacytomas Development of hypercalcemia >11.5 mg/dL related to plasma cell dyscrasia |
In recent years, the fraction of patients achieving deep responses, including complete remission, after initial MM therapy has increased significantly. This correlates with improved progression-free survival (PFS) and overall survival (OS) in several studies (10). With a deepened level of remission, more sensitive methods to assess and monitor minimal residual disease (MRD) have been investigated. These include flow cytometry, allele-specific polymerase chain reaction (ASO-PCR), and next-generation sequencing (NGS)–based assays (11). MRD may soon be used as a valid surrogate end point to compare treatment strategies and advise on consolidation and maintenance therapies. At present, MRD assessment by multiparameter flow cytometry is the most reproducible method in MM. It has a sensitivity of 10–5 if at least 2 × 106 cells from bone marrow aspirates are analyzed. An international effort to adopt standardized or harmonized MRD detection assay and analysis by multiparameter flow cytometry in clinical practice and in clinical trials is ongoing.
After the diagnostic workup and risk stratification are complete, patients who meet the criteria for MM as defined by IMWG criteria should initiate therapy. The most important initial assessment is whether a patient is a candidate for high-dose chemotherapy and autologous stem cell transplantation (SCT), largely based on existing comorbidities and age. In the transplant-eligible population, current MM standard of care involves frontline chemotherapy, followed by consolidative high-dose melphalan and autologous SCT, followed by maintenance therapy. Some chemotherapy agents (eg, melphalan) may adversely affect stem cell collection and should be avoided in the initial therapy of transplant-eligible patients. Melphalan may be included in the frontline therapy of transplant-ineligible patients.
A number of different regimens can be used in the frontline setting for transplant-eligible patients. These usually consist of two- or three-drug combinations, and the choice of therapy is individualized based on factors such as patient comorbidities (neuropathy, diabetes), preferred route of administration (oral, intravenous, or subcutaneous), and underlying disease biology [eg, bortezomib in t(4;14) and del 13q]. Patients are usually given two to four cycles of therapy prior to stem cell collection to reduce disease burden before proceeding to high-dose chemotherapy and autologous stem cell rescue. Given the evidence that the depth and duration of response may translate into improved long-term outcomes, we generally prefer the three-drug regimens over the two-drug regimens as initial therapy in newly diagnosed patients.
The efficacy of the second-generation immunomodulatory drug (IMiD) lenalidomide combined with dexamethasone (Len/Dex) was initially demonstrated in the relapsed and refractory setting. Subsequently, a randomized phase III study in newly diagnosed MM compared lenalidomide plus high-dose dexamethasone versus placebo plus high-dose dexamethasone (12). Overall response rates (ORR), defined as a partial response or greater, and very good partial response (VGPR) rates were significantly higher in the Len/Dex arm (78% and 63%, respectively) versus the placebo/Dex arm (48% and 16%, respectively). The 1-year PFS rate was also higher in the Len/Dex arm (78% vs 52%), although there was no significant difference in OS between the two groups (94% vs 88%). Grade 3 or 4 neutropenia was higher with Len/Dex (21% vs 5%), as was the rate of venous thromboembolism (VTEs) (23.5% vs 5%) despite aspirin prophylaxis.
To possibly decrease the dexamethasone dose while retaining efficacy, a randomized study was conducted with lenalidomide in combination with high-dose dexamethasone (40 mg on days 1-4, 8-11, and 17-20 every 4 weeks) versus low-dose dexamethasone (40 mg on days 1, 8, 15, and 22 every 4 weeks) (13). Patients receiving high-dose dexamethasone achieved a higher ORR (79% vs 68%) after four cycles of therapy. However, a second interim analysis at 1 year demonstrated a statistically significant superior OS in the low-dose dexamethasone arm compared to the high-dose arm (96% vs 87%). This was attributed to increased toxicities of high-dose dexamethasone therapy including VTEs and infections. Based on this study, lenalidomide is recommended to be given in combination with low-dose dexamethasone.
The proteasome inhibitor bortezomib in combination with dexamethasone was studied as frontline therapy in a large phase III trial comparing bortezomib and dexamethasone versus vincristine, doxorubicin, and dexamethasone (VAD) therapy prior to autologous SCT (14). Postinduction ORR (78.5% vs 62.8%), ≥VGPR rates (37.7% vs 15.1%), and complete response (CR) or near complete response (nCR) rates (14.8% vs 6.4%) all favored the bortezomib and dexamethasone arm over the VAD arm. There was also a trend toward improved median PFS in the bortezomib and dexamethasone arm (36.0 vs 29.7 months) but no difference in OS. In a separate analysis, initial treatment with bortezomib and dexamethasone prior to autologous SCT was shown to overcome the adverse prognostic features of t(4;14) in relation to event-free survival (EFS) and OS compared to VAD, although del 17p remained a poor prognostic factor regardless of the treatment regimen. Herpes simplex prophylaxis with acyclovir or valacyclovir should be given with bortezomib-containing regimens. Subcutaneous administration of bortezomib is preferred because it has similar efficacy as the intravenous route with decreased peripheral neuropathy (15).
The addition of oral cyclophosphamide to bortezomib and dexamethasone (CyBorD) was studied in phase II trials. In the EVOLUTION phase II trial, patients were randomized to receive bortezomib, lenalidomide, and dexamethasone (VRD); bortezomib, lenalidomide, cyclophosphamide, and dexamethasone (VDCR); or CyBorD, all followed by maintenance bortezomib for four 6-week cycles (16). The study was later amended to add an additional day 15 dose of cyclophosphamide, in addition to days 1 and 8, in patients receiving CyBorD. Patients receiving the modified CyBorD regimen achieved an ORR of 82%, with a VGPR or better rate of 53% and a CR rate of 47%. The 1-year PFS rate was 100%.
In another phase II study, standard twice-weekly (days 1, 4, 8, and 11) bortezomib was compared to weekly bortezomib (days 1, 8, 15, and 22) in combination with weekly cyclophosphamide and dexamethasone (17). ORR (88% vs 93%) and VGPR rates (60% vs 61%) were similar in both the twice-weekly and weekly bortezomib groups. In addition to demonstrating the efficacy of the three-drug combination of CyBorD, this study also suggested that weekly (instead of twice-weekly) bortezomib could be used to reduce treatment-related toxicity because it resulted in fewer grade 3 and 4 adverse events compared to the twice-weekly schedule (37% and 3% vs 48% and 12%, respectively).
The efficacy of VRD has also been demonstrated in several phase II trials. A phase I/II study evaluating the safety and efficacy of VRD resulted in an impressive 100% ORR in the phase II part, with 74% of patients achieving VGPR or better (18). As mentioned, VRD was also included as one of the arms in the phase II EVOLUTION trial, which resulted in an 85% ORR, with a VGPR or better rate of 51% and a CR rate of 24% (16). Phase III studies are ongoing comparing VRD with bortezomib and dexamethasone (NCT00522392) or lenalidomide and dexamethasone (NCT00644228) in the frontline setting. In addition, the role and timing of autologous SCT are being reexamined in the era of novel agents in a large international phase III trial of frontline VRD followed by lenalidomide maintenance therapy (with the option of SCT at the time of relapse) versus VRD followed by autologous SCT as per the current standard of care (NCT01208662). Phase II studies are also evaluating the efficacy of the second-generation proteasome inhibitor carfilzomib in combination with lenalidomide and dexamethasone (CRD) in the frontline setting with delayed autologous SCT; early results show rapid and deep responses with less peripheral neuropathy (19,20). These studies will clarify the role of novel triplet combinations in the frontline setting and provide insight as to whether deeper responses, including molecular responses, with the multiple novel agents in combination, ultimately translate into improved long-term outcomes (Table 11-6).
| Author | Phase | Treatment (No.) | % ORR (CR) | PFS | OS |
|---|---|---|---|---|---|
| Zonder et al (12) | III | Len + HD Dex (97) HD Dex (95) | 78 (26)a 48 (4) | 1-year: 78%a 1-year: 52% | 3-year: 79% 3-year: 73% |
| Rajkumar et al (13) | III | Len + HD Dex (223) Len + LD Dex (222) | 81 (5)a 70 (4) | 19.1 mo 25.3 moa | 1-year: 87% 1-year: 97%a |
| Harousseau et al (14) | III | VAD (242) VD (240) | 63 (3) 79 (13)a | 29.7 mo 36.0 mo | 3-year: 77% 3-year: 81% |
| Kumar et al (16) | II | CyBorD (33) VRD (42) VRDC (48) CyBorD-mod (17) | 75 (22) 85 (24) 88 (25) 100 (47) | 1-year: 93% 1-year: 83% 1-year: 86% 1-year: 100% | 1-year: 100% 1-year: 100% 1-year: 92% 1-year: 100% |
| Richardson et al (18) | II | VRD (35) | 100 (37) | 18-month: 75%b | 18-month: 97%b |
Initial treatment regimens for transplant-eligible patients can also be used in transplant-ineligible patients. Without the need to collect autologous stem cells, the alkylating agent melphalan can be incorporated into frontline therapy in nontransplant candidates. For 40 years, melphalan and prednisone (MP) represented the standard of care for transplant-ineligible patients. However, the addition of novel agents to the MP backbone and non–melphalan-containing combinations now form the new standard of preferred regimens.
The Gruppo Italiano Malattie Ematologiche dell’Adult (GIMEMA) compared melphalan, prednisone, and thalidomide (MPT) with MP (21). The ORR (76% vs 47.6%) and nCR/CR rates (27.9% vs 7.2%) favored the MPT arm. The median PFS was better in the MPT arm (21.8 vs 14.5 months), although the median OS was similar (45.0 vs 47.6 months). Subsequent phase III studies also demonstrated improved ORR and PFS with MPT compared to MP, with both the Intergroupe Francophone du Myélome (IFM) 99-06 and IFM 01-01 studies also showing a higher OS rate with MPT compared to MP.
Melphalan, prednisone, and lenalidomide (MPL) was also compared with MP alone in a phase III trial comparing MPL with lenalidomide maintenance (MPL-L) versus MPL versus MP (22). The ORR was significantly higher in patients receiving MPL-L or MPL (77% and 68%, respectively) compared to those receiving MP (50%). Although MPT and MPL are superior to MP alone in terms of ORR and PFS, increased toxicity with the addition of a third drug must be carefully balanced with enhanced efficacy, because grade 3 and 4 adverse events were more pronounced in the MPT and MPL arms compared to MP. Although not compared head-to-head, nonhematologic grade 3 and 4 adverse events were less frequent with MPL compared to MPT (22,23).
Bortezomib plus MP (VMP) was also compared with MP alone in a large randomized phase III trial. The ORR and CR rates were 71% and 30%, respectively, in patients receiving VMP versus 35% and 4%, respectively, in the MP arm. Median PFS was better with VMP (24.0 vs 16.6 months). An OS benefit for VMP versus MP (median, 56.4 vs 43.1 months) was also reported in the final analysis (24). Again, the benefits of efficacy must be weighed carefully against toxicity, as grade 3 and 4 adverse events, particularly peripheral neuropathy, were greater in the VMP arm (13%).
The role of melphalan-containing regimens in transplant-ineligible patients has been challenged. Lenalidomide and low-dose dexamethasone (Rd) in four-week cycles until disease progression versus the same regimen for a fixed duration of 72 weeks versus MPT in 6-week cycles for 72 weeks were compared in a randomized phase III study in over 1,500 transplant-ineligible patients (25). Although the ORRs were similar between the three arms, median PFS favored continuous Rd (25.5 months) versus 18 cycles of Rd (20.7 months) and MPT (21.2 months). There was a trend toward improved 3-year OS with continuous Rd (59%) versus fixed-duration Rd (56%) and MPT (51%). There was also a trend toward fewer grade 3 and 4 adverse events in the continuous Rd arm (70%) compared to the MPT arm (78%), in particular grade 3 and 4 neutropenia and neuropathy. However, there was a higher incidence of grade 3 and 4 infections with continuous Rd (29%), likely related to the longer duration of glucocorticoid use.
A community-based phase IIIB trial compared bortezomib and dexamethasone (BD) versus bortezomib, thalidomide, and dexamethasone (BTD) versus melphalan, prednisone, and bortezomib (MPB) followed by maintenance bortezomib (26). The ORR, PFS, and OS were similar across all three arms. Discontinuation due to adverse events was highest in the BTD arm (35%) compared to BD (24%) and MPB (30%). This demonstrates the safety and efficacy of the use of BD in the elderly population. In general, the incorporation of novel agents to combination therapy has improved ORR and long-term outcomes in elderly, transplant-ineligible patients. However, treatment must be individualized based on comorbidities and disease characteristics as well as the patient’s own goals of care (Table 11-7).
| Author | Treatment (No.) | % ORR (CR) | Median PFS (months) | Median OS (months) |
|---|---|---|---|---|
| Facon et al (103) | MPT (125) MP (196) | 76 (13)a 35 (2) | 27.5a 17.8 | 51.6a 33.2 |
Palumbo et al (21
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
