FMTC, MEN-2, and MEN-3 are caused by specific mutations of the RET proto-oncogene.3,4 They are also inherited as an autosomal dominant condition. Patients with FMTC develop MTC usually late in adulthood with no other endocrine tumors. The MTC in this condition is indolent and nonmetastatic such that patients typically die from other causes. MEN-2 (also called MEN-2a) is characterized by medullary thyroid cancer (MTC), pheochromocytoma, and parathyroid hyperplasia. Cutaneous lichen amyloidosis has been described in several kindreds. MEN-3 (also called MEN-2b) is also associated with a RET mutation, but it is in the intracellular tyrosine kinase domain of the molecule. These patients develop a more aggressive form of MTC at a young age, and they have characteristic eye and bony changes. They also have bilateral pheochromocytomas usually in adulthood, but they do not have primary hyperparathyroidism (HPT). Finally, MEN-4 (also called MENX) is caused by a mutation in CDKN1B that encodes p27Kip1, a tumor suppressor gene that inhibits cell cycle progression.5 These patients may be confused with MEN-1 because they develop pituitary and parathyroid adenomas.6 This chapter will further characterize and describe these syndromes including specific molecular and genetic changes, epidemiology, screening, malignant potential, causes of death, and treatment.
MULTIPLE ENDOCRINE NEOPLASIA TYPE 1
Incidence and Etiology in Multiple Endocrine Neoplasia Type 1
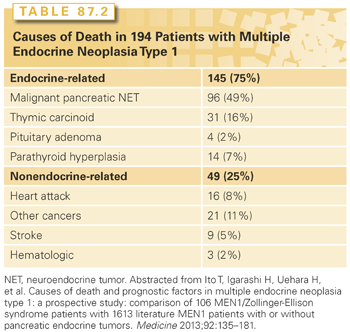
Patients with MEN-1 develop parathyroid hyperplasia and primary HPT typically as the first manifestation of the syndrome (90% to 100%),1,2 followed by pancreatic neuroendocrine tumors (pNET) either functional (20% to 70%) or nonfunctional (80% to 100%), pituitary adenomas (20% to 65%), adrenal tumors (10% to 73%), and thyroid adenomas (0% to 10%). Patients with MEN-1 also have a high occurrence of other endocrine and nonendocrine tumors including carcinoid tumors (thymic 0% to 8%, gastric 7% to 35%, bronchial 0% to 8%, and infrequently intestinal), skin and subcutaneous tumors (angiofibromas 88%, collagenomas 72%, lipomas 34%, and melanoma), central nervous system tumors (meningiomas, ependymomas, schwannomas 0% to 8%), and smooth muscle tumors (leiomyomas and leiomyosarcomas 1% to 7%). The main causes of death in these patients are the malignant potential of the pNET followed by the thymic carcinoid tumors2 (Table 87.2).
Molecular and Genetic Basis in Multiple Endocrine Neoplasia Type 1
MEN-1 is inherited as an autosomal dominant disorder and has an incidence between 0.22% and 0.25% in autopsy studies. The gene causing MEN-1 is located on the long arm of chromosome 11 (11q13).7 It encodes a 610 amino acid nuclear protein called menin that controls cell division, genomic stability, and transcriptional regulation. It is a tumor suppressor gene. Abnormalities of the gene can result in mutations, deletions, and truncations of menin protein. Menin acts as a scaffold protein and increases or decreases gene expression by epigenetic regulation via histone methylation. It complexes with trimethylate histone H3 at lysine, which subsequently facilitates activation of transcription in cyclin-dependent kinase inhibitors and silences transcriptional activity in other target genes. MEN-1–associated tumors harbor germline and somatic mutations.
Screening in Multiple Endocrine Neoplasia Type 1
Genetic screening for MEN-1 is recommended when an individual has two or more MEN-1–related tumors, multiple abnormal parathyroid glands before age 30 years, recurrent HPT at a young age, gastrinoma and HPT or multiple pNETs at any age, plus a family history of kidney stones or endocrine tumors that are part of the syndrome.8 Genetic testing includes sequencing of the entire coding region of the MEN-1 gene (exons 2 to 10) and identifies mutations in about 80% of patients with familial MEN-1.
Primary Hyperparathyroidism in Multiple Endocrine Neoplasia Type 1
Primary HPT is the most common endocrine abnormality in MEN-1. It reaches nearly 100% penetrance by the age of 50 years. HPT is usually the first manifestation of MEN-1 with a typical age of onset of 20 to 25 years. Decreased bone density and kidney stones are common. HPT often occurs at the same time as Zollinger-Ellison syndrome (ZES) and surgery to correct the HPT greatly ameliorates the clinical findings of ZES.9 As in sporadic cases, biochemical testing for HPT is critical to the diagnosis. Total or ionized serum level of calcium and intact serum parathyroid hormone levels are measured and both should be elevated. Twenty-four-hour urinary calcium should also be measured and will be elevated. Patients with MEN-1 and HPT typically have multiple abnormal glands. The tumors are asymmetric in size and should be considered as independent clonal adenomas.8 Imaging studies are not useful for initial operations because all four parathyroid glands must be identified. The current operation of choice is a subtotal parathyroidectomy (3.5 gland resection) with removal of the thymus. Intraoperative parathyroid hormone level monitoring is recommended to be certain that sufficient abnormal parathyroid tissue has been removed. A viable 50 mg amount of normal parathyroid tissue should be left in the neck and marked with a hemoclip. Because of the multiple abnormal parathyroid gland nature of this disease, there is a high probability of recurrent HPT years after surgery if <3.5 resections are performed. Calcium-sensing receptor agonists (calcimimetics) are a new class of drugs that can act directly on the parathyroid gland, decrease parathyroid hormone release, and may even decrease parathyroid tissue growth. These agents may play an important role in the management of these patients in the future.8
Enteropancreatic Neuroendocrine Tumors in Multiple Endocrine Neoplasia Type 1
The prevalence of enteropancreatic NETs in MEN-1 is between 30% to 75%.1,2 The enteropancreatic pathology in MEN-1 is typically multicentric and multifocal with multiple endocrine tumors throughout the pancreas and the intestine. Tumors vary from microadenomas to carcinomas with lymph node and liver metastases. Duodenal gastrinomas in MEN-1 are usually small (<1 cm), submucosal, and multifocal. pNETs contain in decreasing frequency chromogranin, pancreatic polypeptide, glucagon, insulin, proinsulin, somatostatin, gastrin, vasoactive intestinal polypeptide, serotonin, calcitonin, growth hormone releasing factor, and neurotensin. Malignant enteropancreatic NETs are rare prior to the age of 30 years; however, 50% of middle-aged patients with MEN-1 have evidence for malignant pNETs. Recent studies identified the following factors as suggestive of poor prognosis: higher fasting serum levels of gastrin, presence of more than one functional hormonal syndrome, need for more than three parathyroid surgical procedures, presence of either liver metastases, aggressive primary tumor growth, large pNETs (>4 cm), pNETs with areas of poor vascular enhancement on computed tomography (CT), and serial imaging with evidence for progression.2
Gastrinomas (ZES) are the most common functional NET in MEN-1.10 Approximately 40% of patients with MEN-1 will have ZES, and 25% of all ZES cases occur in patients with MEN-1. ZES is diagnosed by elevated fasting serum level of gastrin (>100 pg/ml, off proton pump inhibitors) and concomitant increased gastric acid output (>10 meq/hr). Approximately one-third of patients with ZES will die from the malignant progression of the tumor. Correlates for a poor prognosis with MEN-1/ZES are pancreatic primary tumors, metastases, Cushing syndrome, and severe hypergastrinemia (defined as >3,000 pg/ml).2 Surgery has been done to try to cure patients with MEN-1 of ZES. Local removal of tumors without duodenal/pancreatic resection are associated with persistent ZES; however, Whipple pancreaticoduodenectomy is more consistently associated with biochemical cure.11 This approach is still controversial because those who favor less than Whipple argue that the symptoms of ZES are well controlled with proton pump inhibitors and the morbidity/mortality of Whipple procedure is too significant.
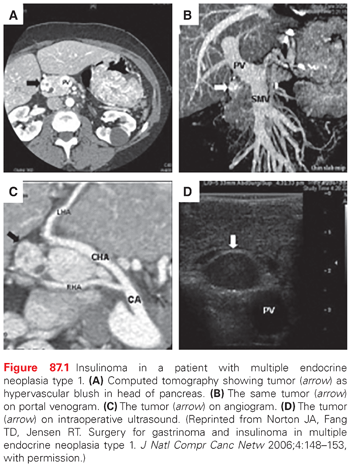
Insulinoma is the second most common functional pNET in MEN-1.12 Approximately 10% of patients with MEN-1 will develop insulinoma, and 10% of insulinomas occur in the setting of MEN-1. Patients have hypoglycemia and neuroglycopenic symptoms (altered mental status and seizures). This occurs at a young age (<35 years). Fasting hypoglycemia (glucose <45 mg/dl) and concomitant hyperinsulinemia (levels >5 uU/ml) are diagnostic. Tumors are generally small (<2 cm) and distributed uniformly throughout the pancreas.1,8 Since patients with MEN-1 have multiple pNETS, it may not be clear which tumor is secreting the excessive insulin. However, one study reported that the insulinoma is most commonly a dominant pNET that is easily identified by conventional imaging studies like CT or magnetic resonance imaging (MRI) 12 (Fig. 87.1).
For nonfunctional enteropancreatic NETS in asymptomatic patients with MEN-1, there is controversy over the role of surgery. Several groups advocate that surgery should be avoided unless the tumor is 2 cm or growing.13 Other groups recommend surgery for tumors that are 1 cm.14 The goal of surgery in MEN-1 appears to be control tumor to prevent cancerous progression. The standard operation is distal or subtotal (resection margin superior mesenteric vein) pancreatectomy with intraoperative ultrasound and enucleation of tumors from the pancreatic head and duodenum. Extensive pancreaticoduodenal procedures are associated with increased risk, thus the indication for the procedure, potential benefit, and surgeon experience must be considered.
Pituitary Tumors in Multiple Endocrine Neoplasia Type 1
Anterior pituitary adenomas are the initial clinical manifestation of MEN-1 in 25% of cases.2,8 Its prevalence in MEN-1 is between 10% and 60%. Most anterior pituitary tumors are microadenomas (<1 cm in diameter). Every type of anterior pituitary tumor has been reported to occur in MEN-1 with the most common type being a prolactinoma. Screening for anterior pituitary tumors requires measuring serum levels of prolactin, insulin-like growth factor 1, and MRI of the pituitary. Patients should be questioned for loss of peripheral vision and visual fields assessed formally if any suspicion. Treatment of pituitary tumors in MEN-1 is the same as sporadic pituitary tumors.
Less Common Tumors in Multiple Endocrine Neoplasia Type 1
Other primary sites of MEN-1 tumors include thymus, bronchus, and stomach. The most worrisome are thymic carcinoids because they are the second most common cause of death in patients with MEN-1.2 Thymic carcinoids are seen more commonly in men with MEN-1 and are usually asymptomatic. Bronchial carcinoids also occur at a higher frequency and are more common in women with MEN-1. In patients with MEN-1 with ZES who have been taking proton pump inhibitors for a long time period, type II gastric enterochromaffin-like cell carcinoid tumors have been identified on upper gastrointestinal endoscopy. The tumors are usually multiple and may involve the entire stomach. These tumors may spread to lymph nodes and liver and may require total gastrectomy.15 Each of these three foregut carcinoid tumors in MEN-1 has malignant potential, and physicians caring for these patients should be vigilant for early diagnosis in patients with known MEN-1.
Adrenal cortical tumors are common in MEN-1. The majority are bilateral, hyperplastic, and nonfunctional. Pathology may include cortical adenoma, diffuse hyperplasia, nodular hyperplasia, and carcinoma. Some patients may present with Cushing syndrome secondary to an adrenal tumor, but adrenocorticotropic hormone from a bronchial carcinoid or a pituitary adenoma may also cause hypercortisolism.16
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


