Visit the Endocrine Hypertension: From Basic Science to Clinical Practice , First Edition companion web site at: https://www.elsevier.com/books-and-journals/book-companion/9780323961202 .



Introduction
Primary hypertension, or hypertension without an identifiable secondary cause, is the most common form of arterial hypertension. Over 900 genetic variants have been associated with blood pressure as a polygenic trait [ ]. In addition, environmental factors contribute to the development of primary hypertension. In contrast, rare forms of hypertension are monogenic, i.e., hypertension associated with single genetic alterations that follow a Mendelian pattern of inheritance. Patients with monogenic hypertension typically present with early-onset, severe, or resistant hypertension, potentially with a positive family history [ ]. However, family studies have revealed incomplete penetrance in some cases, with carriers of disease variants showing milder phenotypes or even normotension [ ]. Similarly, electrolyte abnormalities typically associated with some forms of monogenic hypertension may not be present in all mutation carriers.
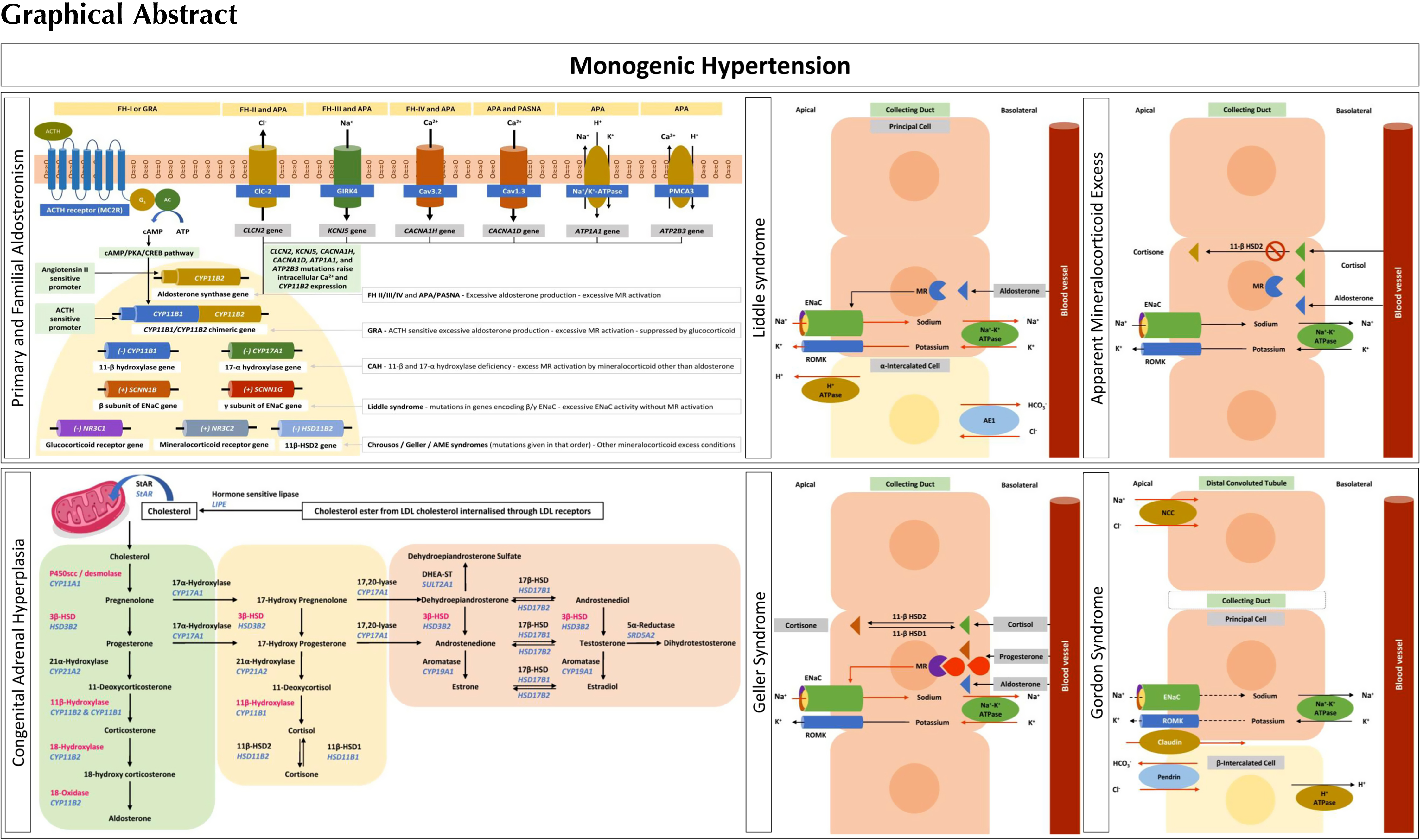
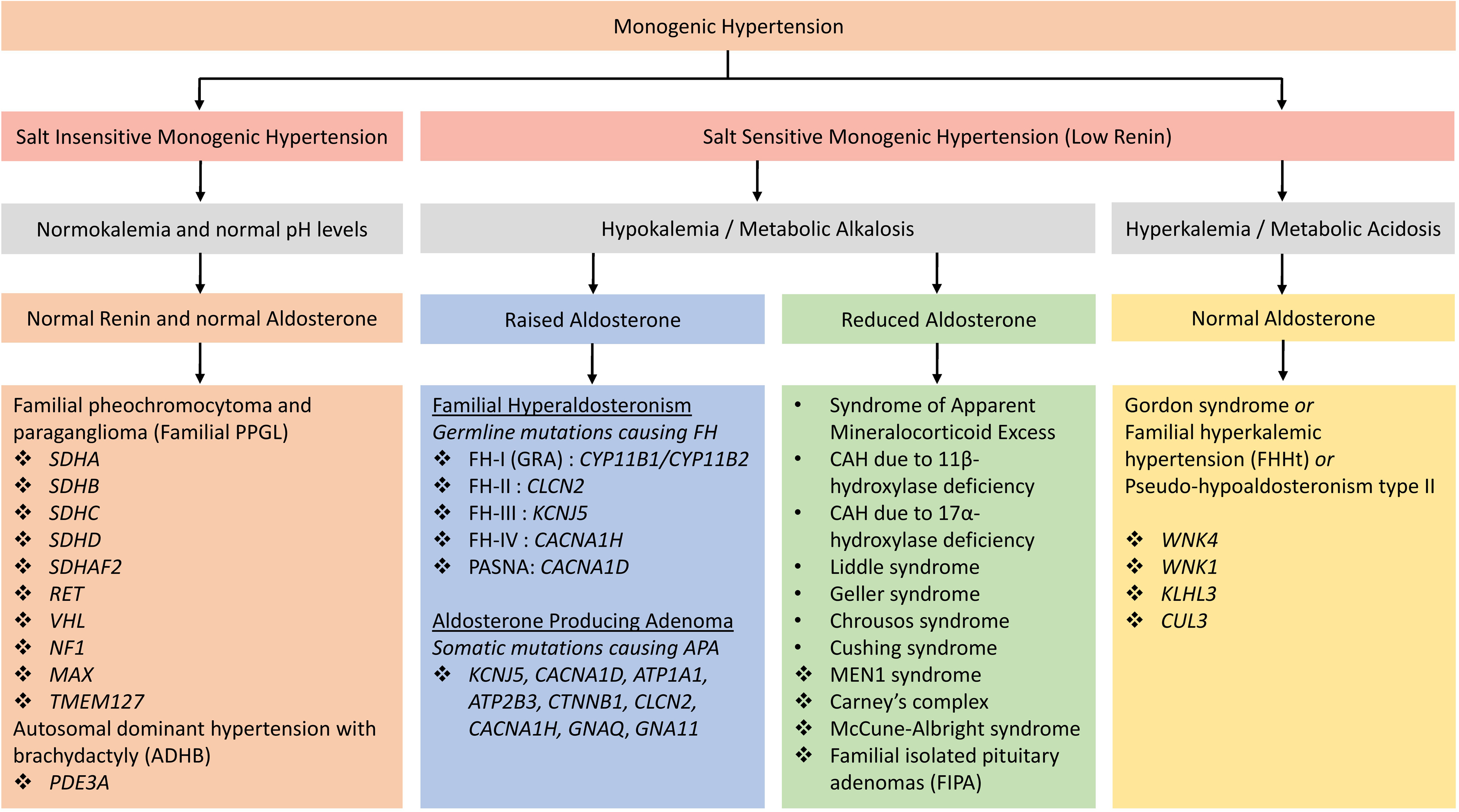
The majority of known monogenic forms of hypertension directly or indirectly lead to increased renal salt reabsorption, plasma volume expansion, and, as a result, suppressed renin levels. Renin thus represents a suitable screening parameter in untreated individuals in whom monogenetic hypertension is suspected. Interpretation can, however, be complicated by antihypertensive medications raising or lowering renin levels. Exceptions with typically normal renin levels include hypertension with brachydactyly (previously known as autosomal dominant hypertension with brachydactyly or ADHB) and familial pheochromocytoma–paraganglioma (see below).
Monogenic hypertension with low renin levels is often associated with characteristic electrolyte abnormalities: either hyperkalemic metabolic acidosis or hypokalemic metabolic alkalosis, although their expression is variable. Gordon syndrome, also known as pseudohypoaldosteronism type II (PHAII) or familial hyperkalemic hypertension (FHHt), is the only monogenic form of hypertension characterized by hyperkalemic metabolic acidosis associated with low renin and normal aldosterone levels [ ]. Subforms associated with hypokalemic alkalosis can again be subdivided into those with increased
Majority of monogenic hypertension syndromes are due to gain-of-function or loss-of-function mutations resulting in altered mineralocorticoid, glucocorticoid, or sympathetic pathways [ ], or changes in renal salt reabsorption and its regulation. The genetic mutations associated with monogenetic hypertension identified kidneys and adrenal glands as the most important players in the regulation of blood pressure [ ].
Monogenic hypertension should be considered in patients with otherwise unexplained childhood-onset hypertension and patients with a family history of early-onset or severe hypertension associated with either low or high potassium levels. Suppressed renin in such patients should trigger further investigations, including aldosterone levels. Associated syndromic features (such as brachydactyly) should similarly trigger genetic investigations. However, mild hypertension and absence of electrolyte abnormalities do not exclude monogenic hypertension, and therefore, hormonal studies should be coupled with genetic testing if monogenic hypertension is suspected. Fig. 6.1 provides an overview and classification of monogenic hypertension. Of note, response to certain medications is increasingly replaced by genetic testing for diagnosis.
Genetics of monogenic hypertension
Monogenic hypertension with hypokalemic alkalosis, low renin, and raised aldosterone
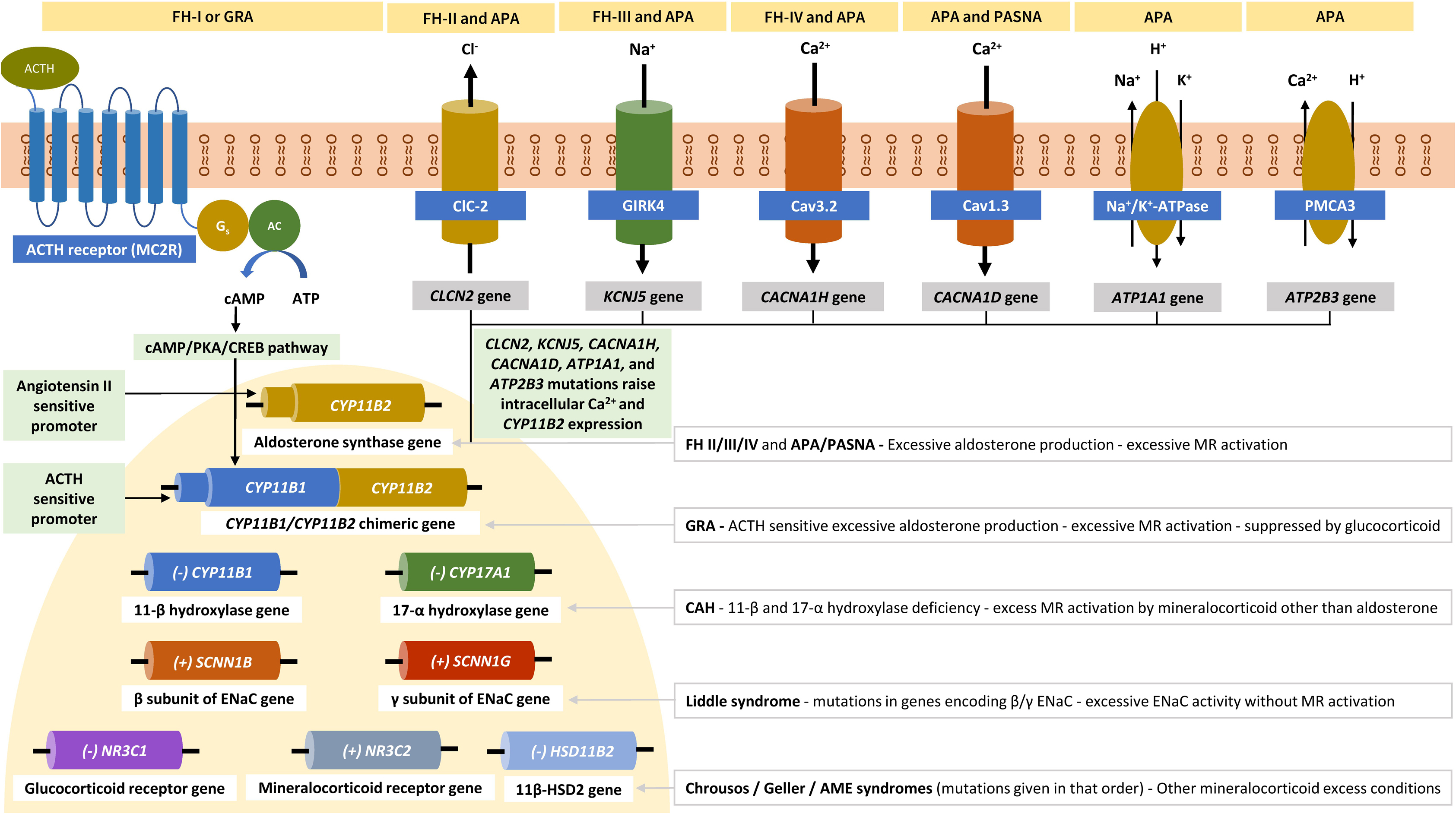
Primary aldosteronism (PA) associated with autonomous aldosterone production by the adrenal gland is the most frequent cause of endocrine hypertension. It has a prevalence of around 6% among hypertensive patients in primary care and around 10% among those referred to hypertension centers [ , ]. The two major causes of PA are aldosterone-producing adenomas (APAs) that account for about 30% of cases and bilateral hyperaldosteronism that accounts for about 60% of cases [ ]. Unilateral adrenal hyperplasia is less common, whereas adrenocortical carcinoma and familial hyperaldosteronism (FH) are both very rare. APAs are typically sporadic. Familial aggregation of APAs has been reported, but most instances are likely chance associations [ ]. More than 95% of sporadic APAs are associated with heterozygous somatic mutations in known disease genes including KCNJ5 , CACNA1D , ATP1A1 , ATP2B3 , CLCN2 , CACNA1H , CTNNB1 , GNA11 , and GNAQ [ ]. Many of these genes encode ion channels or pumps. The KCNJ5 gene, mutated in approximately 40% of APAS, encodes an inwardly rectifying K + channel, GIRK4 [ ]. CACNA1D encodes the voltage-dependent L-type calcium channel subunit alpha-1D (Ca v 1.3) [ ]. ATP1A1 encodes the Na + /K + -ATPase α1 subunit, and ATP2B3 a plasma membrane calcium ATPase (PMCA3) [ ]. CLCN2 encodes the chloride voltage-gated channel 2 (ClC-2) [ ], and CACNA1H encodes the voltage-dependent T-type calcium channel subunit alpha-1H (Ca v 3.2) [ ]. KCNJ5 , CACNA1D , ATP1A1 , ATP2B3 , CLCN2 , and CACNA1H mutations directly or indirectly raise intracellular Ca 2+ levels and thereby increase the expression of the aldosterone synthase gene CYP11B2 , increasing aldosterone production [ ].
CTNNB1 encodes β-catenin involved in the Wnt cell-differentiation pathway [ ]. GNA11 and GNAQ encode G-protein alpha subunits downstream of angiotensin II [ ]. Somatic mutations in these genes confer gain of function.
A causative role of somatic mutations in PRKACA (encoding the cyclic adenosine monophosphate-dependent protein kinase catalytic subunit alpha mutated in cortisol-producing adenomas) [ ] and germline mutations in ARMC5 (encoding armadillo repeat containing 5) in APAs is still debated [ ].
Evidence suggests that race and gender have an impact in the somatic mutation spectrum of APAs. Somatic mutations in the KCNJ5 gene are more common in APAs from Asian populations, whereas those in the CACNA1D gene are more common in patients with recent African ancestry [ ]. Similarly, KCNJ5 and CTNNB1 gene mutations are more frequent in women than in men, whereas CACNA1D and ATP1A1 mutations are more frequent in men. Somatic mutations in genes overlapping with APA disease genes also contribute to the development of aldosterone-producing micronodules as a cause of unilateral or bilateral hyperaldosteronism. Such lesions also occur with advancing age in apparently healthy individuals [ , ].
Contrary to these somatic mutations in sporadic cases, germline mutations cause FH. FH type 1, also known as glucocorticoid-remediable aldosteronism, is an autosomal dominant disease associated with an unequal crossing-over event between CYP11B1 (11-beta-hydroxylase involved in cortisol synthesis) and CYP11B2 (aldosterone synthase), generating a chimeric gene [ ]. The chimeric CYP11B1/CYP11B2 gene leads to ectopic expression of CYP11B2 in the zona fasciculata, with an inappropriate ACTH-dependent regulation of aldosterone biosynthesis. Unlike other genes associated with FH, the chimeric CYP11B1/CYP11B2 gene is not detected in association with sporadic APA [ ]. FH type 2 is an autosomal dominant disease associated with gain-of-function mutation in the CLCN2 gene [ , ]. FH type 3 is an autosomal dominant disease associated with gain-of-function mutation in the KCNJ5 gene [ ]. FH type 4 is an autosomal dominant disease associated with gain-of-function mutation in the CACNA1H gene [ ]. For a more detailed discussion of FH, see Chapter 8 . Fig. 6.2 illustrates various somatic and germline mutations involved in APAs, FH, and other mineralocorticoid disorders associated with monogenic hypertension.
Monogenic hypertension with hypokalemic alkalosis, low renin, and reduced aldosterone
Liddle syndrome is an autosomal dominant disease associated with gain-of-function mutations in the genes for the β, γ, and rarely α subunits of the epithelial sodium channel (ENaC), namely SCNN1B [ ], SCNN1G [ ], and SCNN1A [ ], respectively. In Liddle syndrome with SCNN1B and SCNN1G mutations, these gain-of-function mutations result in the inability to remove ENaC from the cell surface, leading to a constitutively active ENaC associated with excessive sodium transport from the tubular lumen into the interstitium through the sodium/potassium ATPase pump [ ]. The excessive sodium flow thorough the ENaC and the sodium/potassium ATPase pump generates a negative transepithelial voltage, which leads to potassium diffusion through the renal outer medullary potassium (ROMK) channels from the apical membrane of the principal cells to the tubular lumen and facilitates active H + secretion from the apical membrane of the intercalated cells into the lumen with the help of the proton pump. This results in hypokalemic alkalosis. Patients with Liddle syndrome are treated with ENaC inhibitors (amiloride or triamterene). The pathogenesis of Liddle syndrome-mediated hypokalemic alkalosis is shown in Fig. 6.3 .
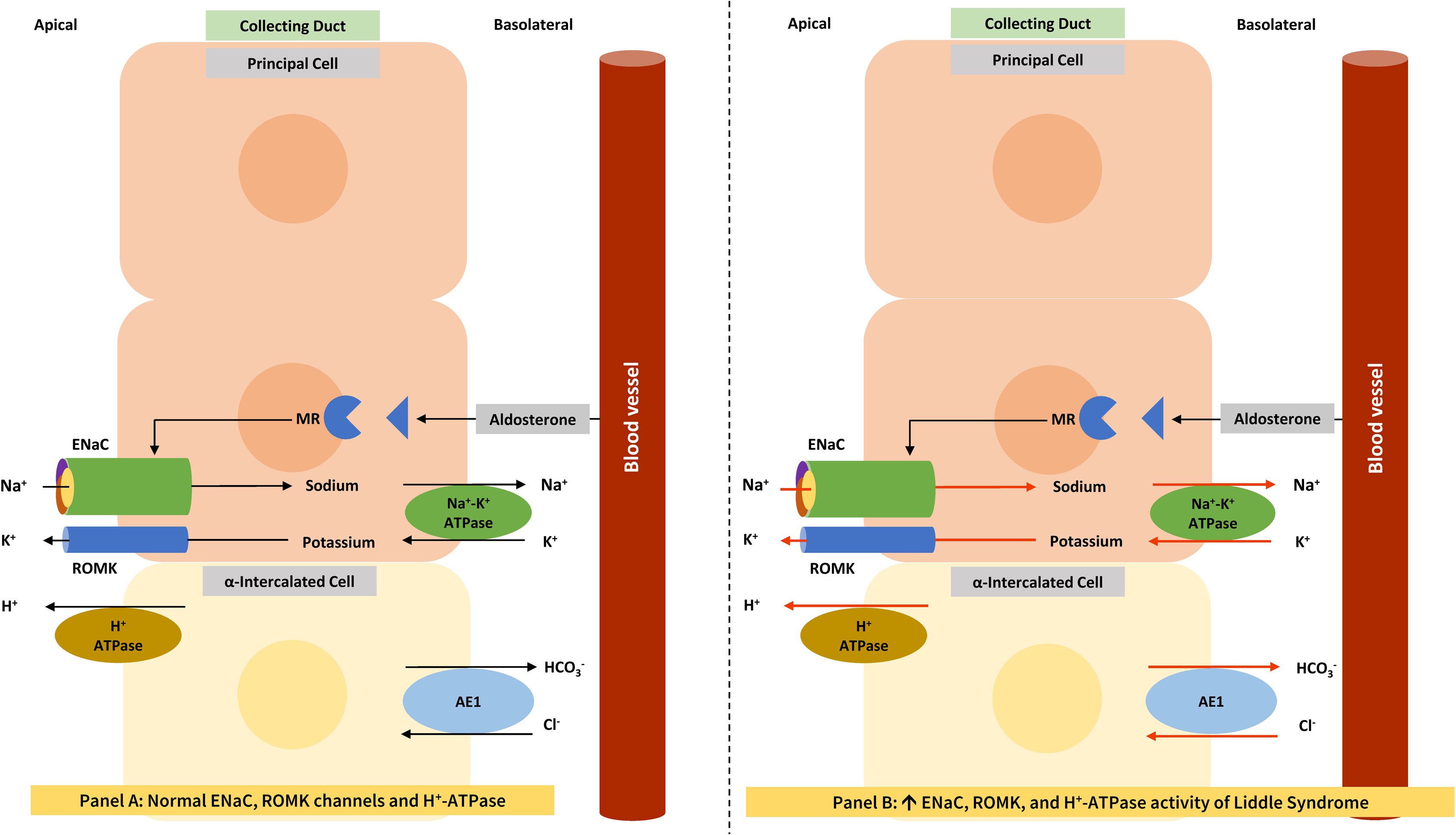
For a better understanding of Figs. 6.3 and 6.5 , the function of the collecting duct is explained in the following paragraph. Though most collecting duct cells are the principal cells (expressing ENaC), different types of intercalated cells including the α-, β-, and non-α/non-β-intercalated cells are also present and contribute to acid base homeostasis [ ]. The acid-secreting α-intercalated cells express H + -ATPase and the Cl − /HCO3 − anion exchanger (AE1) at the apical and basolateral membranes, respectively. In contrast, bicarbonate-secreting β-intercalated cells express the Cl − /HCO3 − anion exchanger (pendrin) and H + -ATPase at the apical and basolateral membranes, respectively. Finally, the non-α, non-β intercalated cells have both pendrin and H + -ATPase at the apical membrane. Pendrin transporters are involved in transcellular chloride reabsorption [ ], whereas claudin channels are involved in paracellular chloride absorption [ ].
Apparent mineralocorticoid excess is an autosomal recessive disease associated with inactivating mutations in the HSD11B2 (hydroxysteroid 11-beta dehydrogenase 2) gene [ ]. The encoded enzyme converts cortisol, which is more abundant than aldosterone and can bind to the mineralocorticoid receptor, to inactive cortisone. Loss of enzyme function results in abnormal mineralocorticoid receptor activation, hypertension, and hypokalemia. Unlike patients with Liddle syndrome, patients with AME respond to treatment with the mineralocorticoid receptor blockers spironolactone and eplerenone. CAH resulting from 11β-hydroxylase deficiency is an autosomal recessive disease associated with inactivating mutations in CYP11B1 gene. CAH due to 17α-hydroxylase deficiency is an autosomal recessive disease associated with inactivating mutations in CYP17A1 gene. Both are infrequent subforms of CAH associated with accumulation of mineralocorticoids, which cause hypertension. Glucocorticoid resistance syndrome ( Fig. 6.4 ) is an autosomal recessive or dominant disease associated with inactivating mutations in the glucocorticoid receptor gene ( NR3C1 ), associated with increased cortisol and mineralocorticoid levels. As these topics are discussed in detail in other chapters of this textbook, we are avoiding repetition in this chapter.