Monoclonal Antibodies in Cancer Treatment
Monoclonal antibodies are used in five different ways in the treatment of human conditions. First, antibodies have a variety of effector mechanisms that focus an array of immunologic agents (complement, various effector cells) on the target to which they bind. Second, antibodies can serve as targeting moieties to specifically deliver diverse killing or inhibitory molecules to a specific site. Third, antibodies can be directed at soluble protein or proteoglycan hormones or cytokines or their receptors to antagonize a particular function such as cell growth, invasion, or migration. Fourth, antibodies can be used as antigens to elicit antitumor responses against immunoglobulin-expressing tumors. Fifth, antibodies can be used to alter the pharmacologic behavior of other substances to either increase or decrease their half-life or alter their distribution (e.g., antibodies to digoxin used to treat digoxin toxicity).
Monoclonal antibody technology was developed in 1975 and has been widely applied in biological sciences since then. The first clinical trial of a monoclonal antibody was performed in 1980 and the first FDA approval of a monoclonal antibody for a cancer indication occurred in 1997. Currently 14 monoclonal antibody-based drugs are FDA-approved for therapeutic use; one monoclonal antibody, nofetumomab (NR-LU10, anti-CD56) labeled with technetium-99m is approved for use as an imaging agent in the staging of small-cell lung cancer (it will not be discussed here). Both the list of agents and their approved uses are likely to expand.
ANTIBODY STRUCTURE AND FUNCTION
Antibody structure was initially elucidated by using antibodies as probes of other antibodies. Three sets of determinants were defined. Isotypes are determinants that distinguish among the main classes of antibodies of a particular species and are defined by antibodies made in different species. Humans have five main heavy chain isotypes (M, G, A, D, E) and two light chain isotypes (κ, λ). Allotypes are small sequence differences or allelic differences between immunoglobulins of the same isotype in different individuals within a species and are defined by antibodies made in the same species. Idiotypes are antigenic determinants formed by the antigen-combining site of an antibody that distinguish each clonal B-cell product.
Antibodies are generally composed of four chains, two identical heavy chains (MW ~50,000 Daltons) and two identical light chains (MW ~22–25,000 Daltons). Each chain has a portion with limited sequence variability called the constant region and a portion with extensive sequence variability called the variable region. The heavy and light chains are linked by disulfide bonds and aligned such that the variable regions of the light and heavy chain are adjacent to each other (Figure 15-1). A specific antigen is bound by the antibody in the pocket formed by the heavy and light chains. The contact regions between the antigen and the antibody are usually defined by two or three regions of hypervariability within the variable regions. These are called complementarity-determining regions (CDRs).
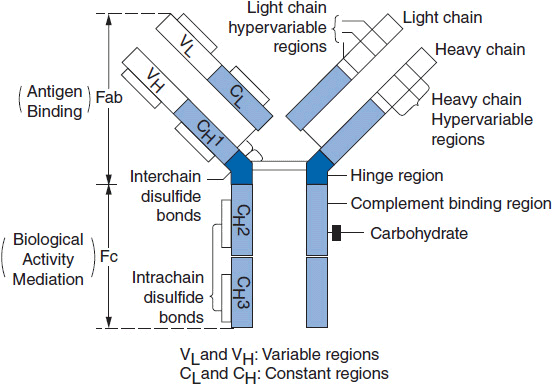
FIGURE 15-1 A schematic depiction of antibody structure and function relationships. (From Wasserman RL, Capra, JD. Immunoglobulins. In MI Horowitz, W Pigman (eds.), The Glycoconjugates, Academic Press, New York, 1977, p. 323.)
It is possible to generate an antibody of defined specificity that can bind to nearly any biological molecule by immunizing mice and isolating and immortalizing the B cell that produces the desired antibody. The B cell is then fused to an immunoglobulin nonproducing B-cell line, yielding the monoclonal murine-derived antibodies first used in clinical trials. The efficacy of murine antibodies was found to be limited by several factors. First, murine antibodies cooperate with human effector mechanisms poorly such that important mechanisms like complement fixation and antibody-dependent cellular cytotoxicity were activated weakly or not at all. Second, the human host has developed sophisticated methods to remove animal proteins rapidly from the blood. Therefore, the biological half-life of murine antibodies is short, indeed, much shorter than the biological half-life of human IgG antibodies (~23 days). Third, murine antibodies are themselves immunogenic. Thus, human antimouse antibodies to the therapeutic agent result in even more rapid clearance on repeat administration. Other factors that compromised efficacy of early antibody trials were tumor related. Targets were picked that were suboptimal. The target molecule could be shed into the serum and distract the antibody from reaching the cell producing the target. In some cases, target molecules were downregulated such that resistance to the therapeutic antibody emerged.
Many of these problems were addressed in a single technical development; the recombinant production of chimeric antibodies that contained the framework and constant regions of human immunoglobulins with the murine-derived antigen binding portion of the molecule (the variable or hypervariable regions). The first of these chimeric antibodies to gain FDA approval and to become widely used clinically was rituximab, an anti-CD20 antibody. The success of rituximab against lymphoid malignancies derived in large measure from the persistence of the company that owned the rights to it. Based on the rather minor antitumor activity of the murine anti-CD20 antibody, a peer-review process would likely have terminated its clinical development. However, the industrial sponsor took the development a step further and generated a chimeric antibody. That final step corrected nearly all of the defects of the murine antibody and pointed the way to other effective antibodies for clinical use.
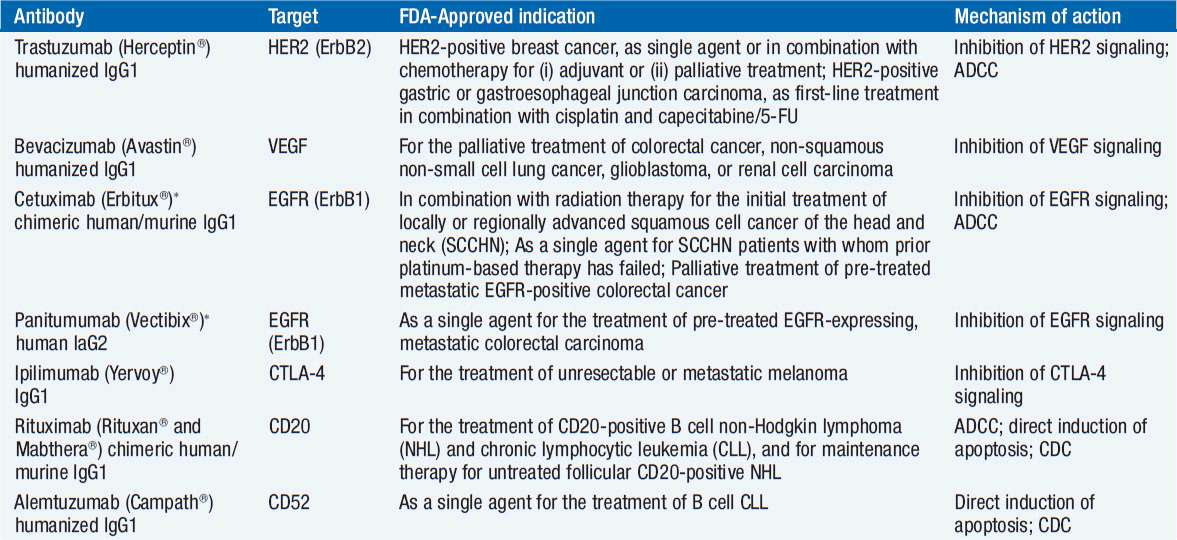
The 14 monoclonal antibodies approved for use in patients with cancer are directed at 8 different targets, CD20 (rituximab, ofatumumab, tositumomab, ibritumomab tiuxetan), epidermal growth factor (EGF) receptor (cetuximab, panitumumab), HER2/neu (trastuzumab, pertuzumab, trastuzumab emtansine), CD33 (gemtuzumab ogomycin), vascular endothelial growth factor (bevacizumab), CD52 (alemtuzumab), CD30 (brentuximab vedotin), and CTLA4 (ipilimumab) (Table 15-1). Antibodies aimed at dozens of potential targets are in development.

RITUXIMAB (RITUXAN)
CD20, the target of rituximab, is expressed mainly on normal and neoplastic B cells. CD20 is a hydrophobic transmembrane protein of molecular weight 35 kD. CD20 is not expressed on hematopoietic stem cells, pro-B cells or plasma cells, or nonlymphoid tissues. The function of CD20 is unclear; some data have suggested that it functions as a calcium channel. It is not shed or internalized upon antibody binding.
Rituximab is a chimeric IgG1, κ antibody with human constant regions and murine variable regions. Its molecular weight is about 145 kD and it binds CD20 with an affinity of 8 nM. Its antitumor effects are thought to be related to its activation of complement and antibody-dependent cellular cytotoxicity. In addition, signaling through CD20 may activate apoptosis mechanisms. Anti-CD20 improves the antitumor effects of chemotherapeutic agents.
The pharmacokinetics of the agent are influenced by a variety of factors including the tumor burden. Early doses tend to achieve lower serum levels because the tumor and normal B cells bind a larger fraction of an administered dose. The empirically derived treatment schedule is weekly doses of 375 mg/m2 IV. After the fourth weekly dose, the half-life averages 205 h with a maximum serum concentration of 486 μg/ml. Levels continue to increase with additional weekly administrations. Delivery of rituximab with chemotherapy does not alter its pharmacology. A maximum tolerated dose has not been defined. Doses as high as 500 mg/m2 are well tolerated. Because of toxicity problems (mainly related to activation of immune effector mechanisms), the drug should be infused at an initial rate of about 50 mg/h.
Toxicities from rituximab are mainly related to the initial infusion. Symptoms generally develop within 30–120 min of starting infusion. In most cases, the symptom complex includes one or more of the following: fever and chills, nausea, pruritis, angioedema, asthenia, headache, broncho-spasm, throat irritation, rhinitis, urticaria, myalgia, dizziness, or hypertension. The reactions resolve entirely with either slowing the infusion or temporarily interrupting it. The infusion-related symptoms generally decrease in incidence with each administration from nearly 80% incidence with the first to around 14% with the eighth. Diphenhydramine, acetaminophen, and intravenous fluids are often required to suppress the symptoms. Once symptoms resolve, the administration of rituximab can be reinitiated at about half the rate of the initial infusion. This symptom complex is thought to be largely due to complement activation. The most severe cases can rarely develop adult respiratory distress syndrome, myocardial infarction, ventricular fibrillation, or cardiogenic shock.
Other uncommon problems include the development of tumor lysis syndrome from rapid killing of tumor cells and occasional Stevens-Johnson syndrome with severe mucocutaneous inflammation. When rituximab is administered with chemotherapy, some patients have experienced reactivation of hepatitis B. In general, rituximab is very well tolerated. It only rarely elicits a host antibody response (~1% of patients). The suppression of normal B cells by rituximab is variable in duration depending on the age of the patient and the length of treatment, but most patients recover normal B-cell function within a year of stopping rituximab. No late effects of B-cell suppression have been reported.
Rituximab is effective in nearly all B-cell-derived malignancies that express CD20. It is particularly active when used in combination chemotherapy and has become a component of standard therapy for diffuse large B-cell lymphoma (see the chapter on non-Hodgkin lymphomas). In addition to its standard use in patients with diffuse large B-cell lymphoma, it is also active in follicular lymphoma, mantle cell lymphoma, chronic lymphoid leukemia, and hairy cell leukemia. It is also being used increasingly to treat autoimmune diseases in which autoreactive antibodies play a pathogenetic role (1). These include idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, autoimmune hemolytic anemia, and some cases of pure red cell aplasia.
OFATUMUMAB (ARZERRA)
Ofatumumab is an IgG1 κ fully human (i.e., not chimeric) monoclonal antibody with specificity for CD20, although it binds an epitope distinct from rituximab. It is approved for use in chronic lymphoid leukemia but is expected to have the same spectrum of antitumor activity as rituximab, given that it attacks the same target. It is thought to kill target cells through complement fixation and activation of antibody-dependent cell-mediated cytotoxicity. Some evidence suggests that it is better at fixing complement and poorer at mediating cellular cytotoxicity than rituximab.
Toxicities include infusion reactions (reduced by premedication with glucocorticoids, acetaminophen, and an antihistamine), neutropenia and thrombocytopenia, infections (especially pneumonia and upper respiratory tract infections), fever, cough, nausea, and diarrhea. The immunosuppression it produces may lead to progressive multifocal leukoencephalopathy or reactivation of hepatitis B.
The agent is cleared mainly by binding to its target. As CD20-bearing cells decrease, its half-life increases. Pharmacokinetics are highly variable, but no adjustments are made based on body weight, age, sex, or creatinine clearance. The antibody is administered as follows: 1. An initial dose of 300 mg; 2. One week later, begin 7 weekly doses of 2000 mg; 3. Four weeks after the seventh weekly dose, give 2000 mg every 4 weeks for 4 doses.
Approval was based on a response rate of 42% among a group of patients with chronic lymphoid leukemia resistant to fludarabine and alemtuzumab (2).
ALEMTUZUMAB (CAMPATH)
CD52, the target of alemtuzumab, is a 21–28 kD cell surface glycoprotein expressed on normal and malignant B and T cells, NK cells, monocytes, macrophages, a subpopulation of granulocytes, as well as a subpopulation of CD34+ bone marrow cells, and on epididymis, sperm, and seminal vesicle, but not on spermatogonia. Its function is unknown. CD52 does not shed or internalize. Alemtuzumab is an IgG1, κ chimeric antibody with human constant and variable framework regions and rat CDRs. It binds to CD52 with a nanomolar affinity and is thought to act through antibody-dependent cellular cytotoxicity.
Alemtuzumab clearance is nonlinear. Its plasma half-life is much shorter for early doses (11 h) than late doses (6 days) presumably because of the depletion of CD52-bearing cells over time. After 12 weeks of doses, the mean AUC is sevenfold higher than the mean AUC after the first dose. No dosage adjustments are required based on age or sex.
Because of infusion-related toxicity, doses are begun at 3 mg/d administered as a 2-h infusion. When infusion-related toxicities are less than or equal to grade 2, the daily dose is escalated to 10 mg. Once that dose is tolerated, one can advance the dose to 30 mg/d. The usual maintenance dose is 30 mg/d 3 times a week, usually a Monday-Wednesday-Friday schedule. Weekly doses exceeding 90 mg total are not recommended because of an increased risk of pancytopenia. Dose escalation from 3 to 30 mg doses can generally be accomplished in a week.
Like rituximab, alemtuzumab is associated with significant infusion-related toxicity with the first dose, decreasing with subsequent administration (3). The symptoms include fever, chills, hypotension, shortness of breath, bronchospasm, and rashes. Rarely the symptoms may progress to adult respiratory distress syndrome, cardiac arrhythmias, myocardial infarction, and heart failure. Routine premedication with diphenhydramine 50 mg and acetaminophen 650 mg 30 min before the infusion is recommended.
The next most common serious toxicity of alemtuzumab is immunosuppression. Because of the widespread expression of CD52 on cells involved in host defenses, patients receiving alemtuzumab become severely immunosuppressed and are susceptible to opportunistic infections such as Pneumocystic carinii, aspergillosis and other fungal infections, and intracellular pathogens like Listeria monocytogenes. The antibody produces profound lymphopenia. CD4+ T-cell counts do not recover above 200/μl for at least 2 months after stopping treatment and full recovery may take more than 1 year. Antiherpes (acyclovir) and anti-infective (bactrim) prophylaxis is recommended and should be continued until lymphocyte recovery. Opportunistic infections may be seen despite prophylaxis. Because of the immune suppression, patients on alemtuzumab who receive blood products should have those products irradiated to prevent graft-vs-host disease. Patients on alemtuzumab should not receive any live vaccines.
The third serious toxicity associated with alemtuzumab is myelosuppression. Neutropenia, anemia, and thrombocytopenia are common, and rarely patients have developed prolonged and occasionally fatal pancytopenia. The mechanism of the cytopenia may be either direct cytotoxicity or autoimmune; idiopathic thrombocytopenic purpura and autoimmune hemolytic anemia have both been documented. Grade 3 or 4 myelosuppression is noted in 50%–70% of patients.
Nearly 2% of patients receiving alemtuzumab generate antibodies to it, but no adverse effects on toxicity or response have been documented.
The main clinical use for alemtuzumab has been as a salvage therapy for chronic lymphocytic leukemia that is unresponsive to alkylating agents and nucleosides. It is being tested as salvage therapy for other lymphomas and is particularly promising in the treatment of T-cell lymphomas. It is being tested as an immunosuppressive agent in graft-vs-host disease and other conditions of immune hyperreactivity and eosinophilia. It is effective at depleting marrow and peripheral blood collections of T cells in vitro before reinfusing the cells in the setting of allogeneic hematopoietic stem cell transplantation.
BEVACIZUMAB (AVASTIN)
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


