Fig. 4.1
Major pathways involved in melanoma. Arrows, activating signals; interrupted lines, inhibiting signals. CDKN2A cyclin-dependent kinase inhibitor of kinase 2A, CDK2/4/6 cyclin-dependent kinase 2/4/6, MDM2 murine double minute 2 (MDM2), ERK extracellular-related kinase, MEK mitogen-activated protein kinase/ extracellular related kinase, MC1R melanocortin-1 receptor, cAMP cyclic adenylate monophosphate, MITF microphthalmia-associated transcription factor, BAD BCL-2 antagonist of cell death, IKK inhibitor-of-kB- protein kinase, PI3K phosphatidylinositol 3-kinase, PTEN phosphatase and tensin homolog
The RAF kinase family consists of three members, ARAF, BRAF, and CRAF, all of which can activate MEK/ERK signaling [11]. In melanoma, the most commonly mutated component of this pathway is the BRAF gene, the prevalent BRAF mutation (in nearly 90 % of cases) being a substitution of valine with glutamic acid at position 600 (V600E) [12]. Mutated BRAF induces constitutive ERK signaling, stimulating proliferation and survival and providing essential tumor growth and maintenance functions [12]. BRAF is implicated in several aspects of melanoma induction and progression, although the presence of BRAF mutations in nevi [13, 14] strongly suggests that BRAF activation is necessary but not sufficient for the development of melanoma.
In a study aimed to better define the role of BRAF in melanomagenesis, a transgenic zebra fish expressing V600EBRAF showed dramatic development of patches of ectopic melanocytes (designated as fish-nevi) [15]. Remarkably, activated BRAF in p53-deficient zebra fish induced the formation of melanocytic lesions that rapidly developed into invasive melanomas that resembled human melanomas in terms of their histology and biological behaviors [15]. These data provide direct evidence that the p53 and BRAF pathways interact functionally during melanomagenesis.
The BRAF gene also cooperates with the cyclin-dependent kinase inhibitor p16CDKN2A (see below). Activating BRAF mutations have been reported to constitutively induce upregulation of p16 CDKN2A and cell cycle arrest (this phenomenon appears to be a protective response to an inappropriate mitogenic signal). In particular, mutant BRAF protein induces cell senescence by increasing the expression levels of the p16CDKN2A protein, which, in turn, may limit hyperplastic growth caused by BRAF mutations [14]. Therefore, inactivation of p16 CDKN2A gene may promote the melanocytic proliferation depending on oncogenic BRAF. In this sense, several factors seem to be able to induce the arrest of the cell cycle and cell senescence caused by BRAF activation [16, 17].
Finally, it has been showed that primary melanomas arising from chronically sun-damaged skin and from mucosal sites, which typically do not harbor BRAF and NRAS mutations, have increased copy number of the CCND1/Cyclin D1 gene [18]. In contrast to primary melanomas, a subset (>15 %) of metastatic melanoma samples with BRAF mutations also exhibit amplification of CCND1/Cyclin D1. These melanomas are resistant to BRAF inhibitors highlighting the need for combination therapy [19, 20].
CDKN2A
The cyclin-dependent kinase inhibitor 2 (CDKN2A; at chromosome 9p21) gene encodes two proteins, p16CDKN2A and p14CDKN2A (a product of an alternative splicing), that are known to function as tumor suppressors [21]. In particular, p16CDKN2A is part of the G1–S cell cycle checkpoint mechanism that involves the retinoblastoma-susceptibility tumor suppressor protein (pRb). The p16CDKN2A inhibits CDK4, which, in turn, phosphorylates pRb and allows progression through the G1–S checkpoint (Fig. 4.1). On the other hand, p14CDKN2A interacts with the murine double minute 2 (MDM2) protein that targets p53 for degradation (Fig. 4.1) [22]. In particular, the p14CDKN2A protein exerts a tumor suppressor effect by inhibiting the oncogenic actions of the downstream MDM2 protein, whose direct interaction with p53 blocks any p53-mediated activity and targets the p53 protein for rapid degradation. Impairment of the p14CDKN2A-MDM2-p53 cascade, whose final effectors are the Bax/Bcl-2 proteins, has been implicated in defective apoptotic responses to genotoxic damage and, thus, to anticancer agents (in most cases, melanoma cells present concurrent high expression levels of Bax/Bcl-2 proteins, which may contribute to further increasing their aggressiveness and refractoriness to therapy) [23].
In normal conditions, expression levels of p53 within cells are low. In response to DNA damage, p53 accumulates and prevents cell division. Therefore, inactivation of the TP53 gene results in an intracellular accumulation of genetic damage which promotes tumor formation [24]. In melanoma, such an inactivation is mostly due to functional gene silencing since the frequency of TP53 mutations is low [25].
The CDKN2A mutations are more frequent in patients with a strong familial history of melanoma (three or more affected family members) relative to patients with no familial occurrence of melanoma [26]. A recent meta-analysis of studies conducted in independent populations indicated that multiple variants of the melanocortin-1 receptor (MC1R) gene increase the melanoma risk in CDKN2A mutation carriers [27].
The MC1R gene encodes a G-protein coupled receptor. In the skin, two types of melanin pigment, dark-protective eumelanin and red photoreactive pheomelanin, are present [28]. MC1R plays an important role in determining the ratio of eumelanin and pheomelanin production (Fig. 4.2). After stimulation by UV, keratinocytes produce alpha melanocyte-stimulating hormone (MSH) that binds to the MC1R on melanocytes and shifts the balance of these two pigments in the direction of eumelanin [28]. In particular, stimulation of MC1R by MSH mediates activation of adenylate cyclase, subsequent elevation of cAMP levels, and activation of the microphthalmia transcription factor (MITF; see below) (Fig. 4.2a). Activated MITF binds to a conserved region found in the promoters of the tyrosinase (TYR), tyrosinase-related protein 1 (TYRP1), and DOPAchrome tautomerase (DCT) genes, stimulating the transcriptional upregulation of these proteins and inducing maturation of the melanosomes [29]. This ultimately results in increased eumelanin production and darkening of the skin or hair.
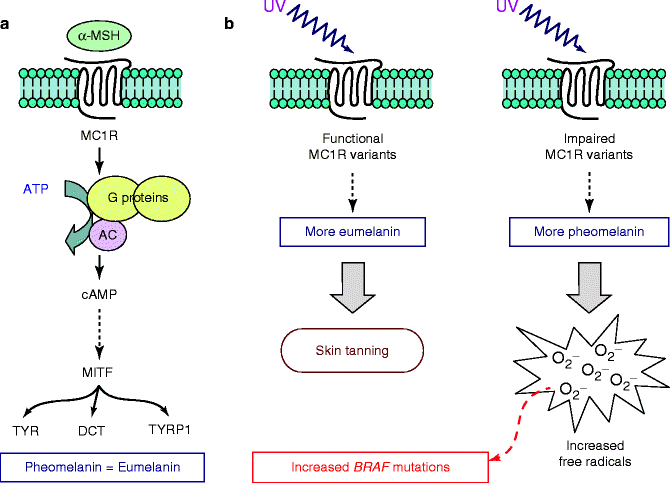
Fig. 4.2
Role of MC1R on pigmentation and melanoma pathogenesis. (a) Functional pathway downstream MC1R, leading to the activation of genes acting as major determinants of hair and skin pigmentation. (b) Effects of the increased production of eumelanin or pheomelanin. αMSH alpha melanocyte-stimulating hormone, TYR tyrosinase, TYRP1 tyrosinase-related protein 1, DCT DOPAchrome tautomerase
New findings have shed light on the mechanisms by which MC1R contributes to melanoma risk. In vitro studies showed that acute UV irradiation of melanocytes with impaired MC1R results in an increased production of free radicals [30]. Melanomas that arise on body sites only intermittently exposed to sun, and which therefore lack marked signs of chronic solar damage, were found to have a high frequency of BRAF mutations [18]. One could speculate that induction of BRAF mutations may occur only when solar exposure is not sufficiently prolonged to induce the striking tissue changes that generate the hallmark signs of solar damage. Several MC1R variants, which are impairing relevant protein function, have been associated with BRAF mutation in melanoma arising in Caucasian populations from the USA and Europe [31–34]. On the basis of such indications, it is possible that increased production of free radicals following UV exposure in combination with impairment of MC1R may induce mutations in the BRAF gene (Fig. 4.2b).
Additional mechanisms promoting susceptibility to pathogenetic mutations of the BRAF gene may however exist since there is no demonstrable association between germ line MC1R status and the prevalence of somatic BRAF mutations in melanomas from Australian population, even after classifying the melanomas by their location relative to intermittent and chronic sun exposure [35].
PTEN-AKT
Phosphatase and tensin homolog deleted in chromosome ten (PTEN) has a key role in cellular signal transduction by decreasing intracellular phosphatidylinositol [3,4-bisphosphate (PIP2) and 3,4,5-trisphosphate (PIP3)] that are produced by the activation of phoshoinosite 3-kinase (PI3K) [36]. Active RAS exerts the strongest stimulus for PI3K activation (Fig. 4.1), resulting in an increase of PIP3 intracellular levels and a consequent conformational change of AKT [37]. Activated AKT in turn phosphorylates its substrate, the serine/threonine kinase mTOR, leading to increased synthesis of target proteins that regulate cell division and apoptosis [37]. The mechanisms associated with the ability of AKT to suppress apoptosis include the phosphorylation and inactivation of many proapoptotic proteins, such as BAD (Bcl-2 antagonist of cell death) and MDM2, as well as the activation of NF-kB [38] (Fig. 4.1).
In primary melanomas, inactivation of PTEN gene is mainly due to hypermethylation-based epigenetic mechanisms, with a low incidence (less than 10 %) of somatic mutations [39]. PTEN inactivation has been mostly observed as a late event in melanoma, although a dose-dependent downregulation of PTEN expression has been implicated in early stages of tumorigenesis [36]. In addition, alterations of the BRAF-MAPK pathway are frequently associated with PTEN-AKT impairment [2, 40]. In summary, the combined effects of the loss of PTEN and activation of the PI3K/AKT pathway may result in aberrant cell growth, apoptosis escape, and abnormal cell spreading and migration.
Microphthalmia-Associated Transcription Factor (MITF)
The microphthalmia-associated transcription factor (MITF) is a basic helix–loop–helix leucine zipper transcription factor that is considered to be the master regulator of melanocyte biology, since it is involved in melanoblast survival and melanocyte lineage commitment. MITF is activated by the MAPK pathway as well as by the cAMP pathway (Fig. 4.1) and leads to transcription of genes involved in pigmentation (TYR, TYRP1, and DCT; see above) as well as cell cycle progression and survival. A genome-wide analysis of copy number alterations in cancer identified MITF as an amplified locus in melanoma; MITF amplification correlated with increased resistance to chemotherapy and decreased overall survival [41].
The connection between MITF and melanoma development is complex because it plays a double role of inducer/repressor of cellular proliferation. High levels of MITF expression lead to G1 cell cycle arrest and differentiation, through induction of the cell cycle inhibitors p16CDKN2A and p21 [42, 43] (Fig. 4.1). Very low or null MITF expression levels predispose to apoptosis, whereas intermediate MITF expression levels promote cell proliferation [41–43]. Therefore, it is thought that melanoma cells have developed strategies to maintain MITF levels in the range compatible with tumorigenesis. It has been shown that constitutive ERK activity, stimulated by V600EBRAF in melanoma cells, is associated with MITF ubiquitin-dependent degradation [44]. Nevertheless, continued expression of MITF is necessary for proliferation and survival of melanoma cells, because it also regulates CDK2 and Bcl-2 genes [45, 46]. It has been recently shown that oncogenic BRAF may control intracellular levels of the MITF protein through a fine balance of two opposite mechanisms: a direct reduction of MITF levels, by inducing protein degradation, and an indirect increase of MITF levels, by stimulating transcription factors which increase protein expression levels [47]. Oncogenic BRAF mutations are associated with MITF amplification in a low fraction (10–15 %) of melanomas [47] suggesting that other mechanisms are likely to be involved in ERK-dependent degradation of MITF.
Nuclear Factor-kB
Nuclear factor-kB (NF-kB) is important in inflammation and cell proliferation and survival [48]. A wide range of stimuli activates NF-kB which, upon activation, translocates to the nucleus to interact with kB sites located in regulatory regions of target genes [49].
Aberrant NF-kB regulation may cause alteration of the transcriptional activation of genes associated with cell proliferation, angiogenesis, tumor promotion, and suppression of apoptosis [50–52]. In melanoma, NF-kB is constitutively activated since expression of the inhibitor-of-kB (IkB) proteins, which form complexes sequestering NF-kB into the cytoplasm, seems to be significantly reduced in comparison to nevi [53]. In particular, activation of the AKT and/or MAPK pathways has been demonstrated to increase degradation of such complexes through activation of an IkB kinase (IKK) effector (Fig. 4.1), with subsequent release of NF-kB which may thus move to the nucleus and activate gene transcription [54–56].
From Molecular Mechanisms to Histopathological Markers
Increased knowledge of molecular mechanisms in melanoma has prompted interest in how this may be applied to improve clinical decision making on the basis of more accurate diagnosis, refined prediction of probable future clinical course (prognostication), and the likelihood of a meaningful response to specific treatment modalities. These assessments currently are based on consideration of patient’s demographics, clinical presentation, and microscopic and immunohistochemical features of the individual melanoma. Clinical assessment relies on elicitation of a detailed clinical history and scrutiny of the skin tumor, possibly with the assistance of a dermoscope. If melanoma is suspected, the lesion is often examined in situ by ultrasound to assess depth of invasion [57]. Standard microscopic evaluation of tissue sections from a primary melanoma, stained by hematoxylin and eosin (HE), classifies the histology and cytology of any melanocytic proliferation present at the dermoepidermal interface, the presence and micrometer-measured extent of ulceration, and the measured thickness and depth of any dermal infiltration by tumor [58]. The tumor cells of junctional and dermal components are evaluated for pleomorphism, nuclear atypia, and the frequency and location of normal and abnormal mitoses. The associated stroma is assessed for the presence, density, and distribution of any peritumoral or intratumoral lymphocytic infiltrates. Evidence of vascular and/or lymphatic invasion sought and neurotropism and angiotropism recorded [59]. Prognosis is based on consideration of the presence and extent of ulceration, the micrometer-measured Breslow thickness, and the frequency of mitoses in the vertical growth phase [60].
Immunohistochemistry
Currently, immunohistochemistry (IHC) is routinely used to differentiate melanomas from benign melanocytic tumors that resemble melanoma in HE-stained tissue sections. We here provide a summary of current practice, but for a detailed account of this complex subject, see Ohsie et al. [61]:
S-100, a 21 kDa acidic calcium-binding protein previously known to be expressed by glial cells, is a sensitive marker for cells of melanocytic lineage that is expressed in the nuclei and cytoplasm of the cells of 97–100 % of melanomas. S-100 is not a specific marker for melanocytic lesions; among others, fibroblasts in dermal scars may express S-100, a potentially major diagnostic pitfall in the evaluation of tissue for desmoplastic melanoma.
HMB45, a marker of the premelanosomal glycoprotein gp100, is not as sensitive as S-100 but is more specific (69–93 %) with greater expression in primary (77–100 %) than in metastatic melanomas (58–83 %). Staining may be patchy or zonal with relatively strong HMB45 expression in the superficial component of primary melanomas and progressively weaker staining deeper. HMB45 may be less sensitive in amelanotic melanomas.
MART-1 (melanoma antigen recognized by T-cells-1) and Melan-A are synonyms for a melanosomal differentiation-associated cytoplasmic protein that is recognized by T-cells. Two antibody clones are available: M2-7C10, generally referred to as MART-1, and A103, generally referred to as Melan-A. These antibodies show sensitivity (75–92 %) and specificity (95–100 %) for melanoma at a similar level to HMB45. Both antibodies stain metastatic melanomas less strongly than primary melanomas. MART-1 and Melan-A are associated with more diffuse and intense cytoplasmic staining than HMB45. They less often show weaker staining of melanoma cells located in the deeper dermis which facilitates interpretation.
Tyrosinase is an enzyme hydroxylating tyrosine as the initial step in melanogenesis. In melanomas, positive staining to tyrosinase is seen as a strong diffuse finely granular pattern in the cytoplasm. The sensitivity of the antibody to tyrosinase for melanoma (84–94 %) exceeds that of HMB45, but sensitivity declines within metastatic lesions (79–93 %). Specificity for melanoma is also high (97–100 %), though tyrosinase is not entirely specific to cells of melanocytic lineage.
MITF (see above) is located in the nucleus and a positive reaction is relatively easy to interpret. Sensitivity for melanoma is 81–100 %, with expression in S-100-negative melanomas. Initial reports claimed 100 % specificity for MITF, but recent reports indicate lesser specificity (88 %), especially in the case of spindle cell melanomas. Reactivity with some non-melanocytic tumors has been reported; this relative non-specificity somewhat offsets the interpretive advantages of nuclear staining.
Cellular proliferation within melanocytic tumors, determined by enumeration of the frequency of mitoses per mm2, may also be assessed using markers of the active phases of the cell cycle, such as Ki67 and PCNA. The general principle is that malignant melanomas have a higher frequency of Ki67-positive nuclei than benign nevi. Additionally, marker-positive nuclei in benign nevi are more frequently located in the superficial area of the lesion. Determination of the frequency of Ki67-positive melanoma cell nuclei can be challenging when there is an abundant population of lymphocytes, which have an inherently high proportion of Ki67-positive nuclei.
Representative examples about the use of the immunohistochemistry in differential diagnosis of melanocytic lesions are shown in Figs. 4.3, 4.4, and 4.5.
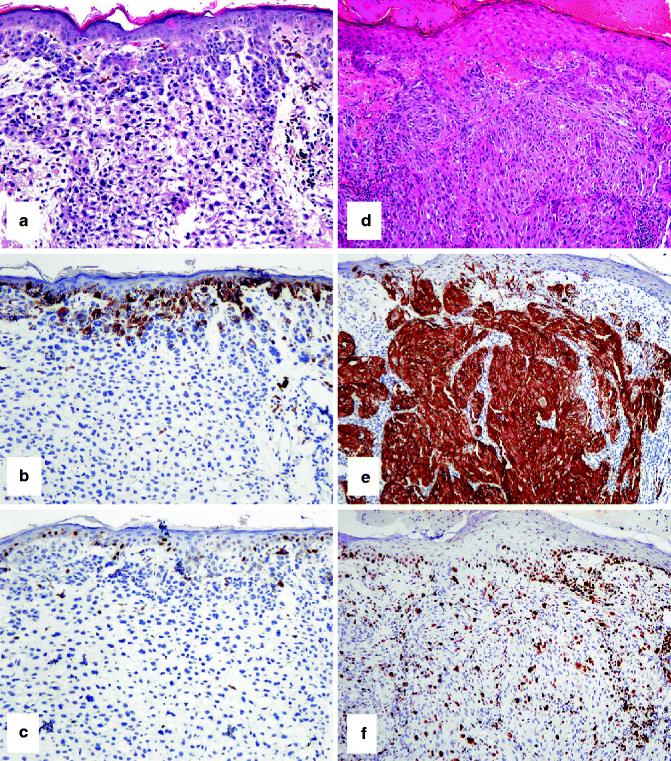
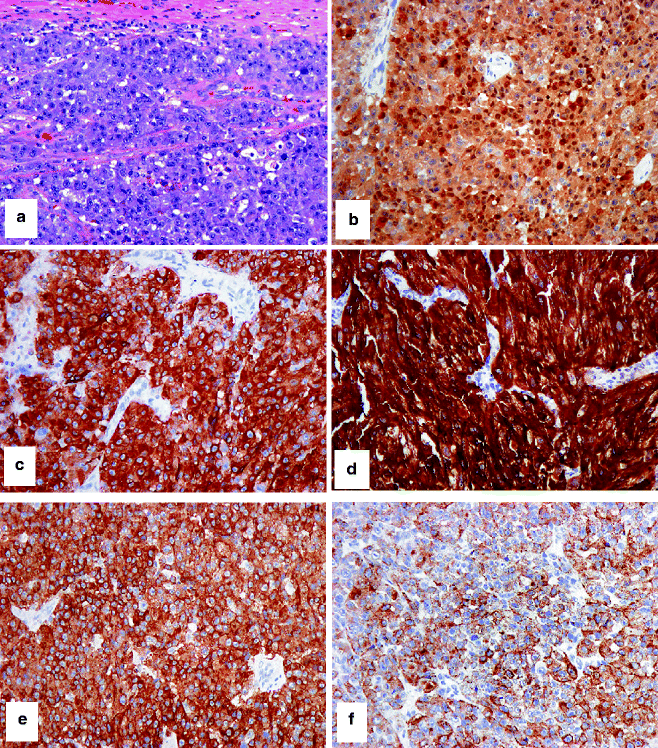
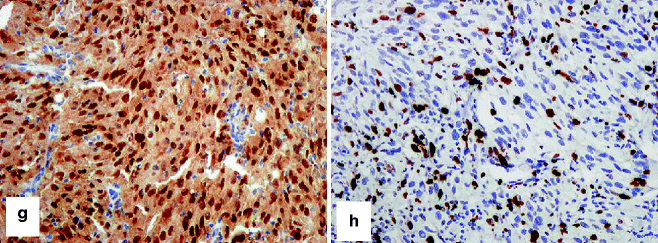
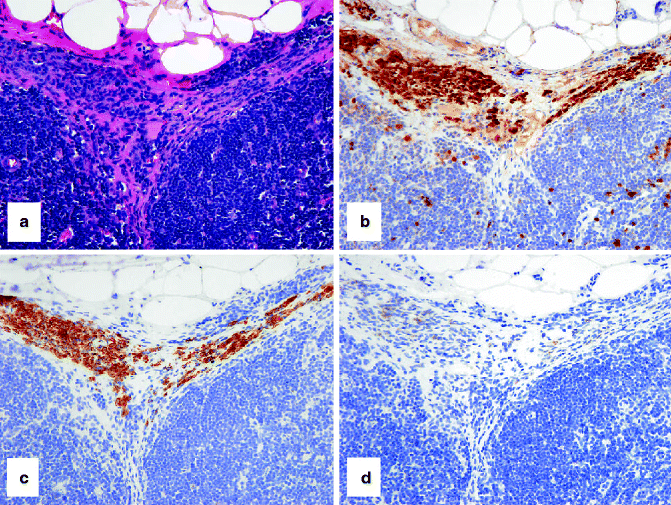
Fig. 4.3
IHC to help separate a compound nevus from a primary melanoma. (a) HE-stained compound nevus that arises at the epidermo-dermal junction and extends into the underlying dermis. (b) Only nevus cells at the junction stain with HMB-45. (c) A similar pattern with Ki67-positive nevus cells (admixed with keratinocytes at the junction but absent from the dermal component). (d) HE-stained primary melanoma arising from the overlying epidermis and extending into the dermis. In contrast to the nevus, (e) strong staining of epidermal and dermal melanoma cells with HMB-45 and (f) frequent junctional and dermal melanoma cells with Ki67-positive nuclei are observed
Fig. 4.4
IHC to confirm that a tumor is melanocytic. (a) HE-stained malignant epithelioid cellular neoplasm, cytologically and histologically suspicious for melanoma, without evidence of melanin synthesis. Differential immunohistochemical studies confirm that the tumor is melanocytic in histogenesis and likely malignant: (b) strong nuclear and cytoplasmic expression of S-100, (c) strong cytoplasmic expression of Mart-1, (d) very strong cytoplasmic expression of tyrosinase, (e, f) variable cytoplasmic expression of HMB-45, (g) strong nuclear expression of MITF, and (h) strong and frequent nuclear expression of Ki67. Note the differential expression of HMB-45 between a primary melanoma (e) and metastatic melanoma (f). This differential epitope expression is often (but not always) present
Fig. 4.5
IHC to identify capsular/trabecular nevus cells in a sentinel lymph node. (a) HE-stained section shows small blue-colored nevus cells in the nodal capsule (pink) and extending downwards in a connective tissue trabeculum. The nevus cells stain strongly with (b) S-100 and (c) MART-1, but weakly or not at all with (d) HMB-45. In contrast, melanoma cells stain strongly with S-100, Mart-1, and HMB-45
These standard clinical and histopathological IHC approaches are time tested, and using such approaches, experienced pathologists can readily and accurately identify more than 95 % of melanomas and separate them from melanocytic nevi. Problems arise in the interpretation of so-called borderline lesions (melanocytic tumors of unknown malignant potential—MELTUMP) [62]. These are tumors in which the histology and/or cytology is typical neither of malignant melanoma nor melanocytic nevus. MELTUMPs include atypical Spitz lesions, atypical cellular blue nevi, deep penetrating nevi, and pigmented epithelioid melanocytomas. There is often very considerable diversity of opinion when such lesions are evaluated by multiple pathologists. In these dilemmas, there is an urgent need for additional accurate diagnostic approaches that will supplement routine clinical and histological evaluation and increase diagnostic accuracy to a level where critical management decisions can be made with confidence.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


