Molecular Pathogenesis of Endometrial Carcinoma
Martin Kobel MD
The collective of endometrial carcinomas is increasingly recognized to comprise a number of distinct diseases.1 The major histologic types of endometrial carcinomas are associated with distinct molecular alterations. Phenotypic criteria, which are used to type endometrial carcinomas, have been defined on an architectural and cellular, nuclear level. These morphologic features are directly related to distinct molecular events that determine the lineage of differentiation and the oncogenic pathway. Serous carcinomas are characterized by anaplastic nuclear features, which correlate with the underlying chromosomal instability, whereas other alterations (e.g., PTEN/PI3K and Wnt/CTNNB1 pathways) are associated with low-grade nuclear features.2
Three decades ago, the first characteristic molecular features for specific types of endometrial carcinomas were reported, with landmark papers describing TP53 mutation in “nonendometrioid” (serous) carcinomas,3 the identification of microsatellite instability in a significant portion of sporadic endometrial carcinomas,4 and PTEN mutation5,6 and CTNNB1 mutation in endometrioid types.7 Initially based on clinical features8 and later on histopathologic and molecular features,9 a binary classification system for endometrial carcinomas has been proposed that divides these tumors into type I (endometrioid, mucinous) and type II (serous, clear cell). This model clearly delineates endometrial carcinomas with respect to their relation to estrogen stimulation. However, the model obscures some of the significant differences within these groups (i.e., between low-grade endometrioid and high-grade endometrioid carcinoma and between serous and clear cell carcinoma). The molecular characterization of endometrial carcinomas has been evolving, and an overview on the mutational landscape across histologic types as known today is given in Table 7-1.10,11,12,13,14,15 and 16
There are four major clinicopathologic variants of endometrial carcinomas that account for more than 95% of cases that differ with respect to molecular alterations, phenotype, biomarker expression, and clinical behavior. These types include low-grade (grade 1 and grade 2) endometrioid, high-grade (grade 3) endometrioid, serous, and clear cell carcinoma.
Most of the alterations shown in Table 7-1 were identified as candidate genes by analogy with tumors from other organ systems. These numbers may represent an underestimate due to the limited sensitivity of conventional Sanger sequencing; however, these case series might also be subject to selection bias. Unbiased comprehensive new genomic and epigenomic analysis is needed to broaden our understanding of the complex molecular basis of these
diseases. Comparison of tumor-type specific mRNA expression profiles has further provided insight into the biologic differences of these distinct tumor types,17,18,19 and 20 and some candidate biomarkers are being used in diagnostic practice (see below).
diseases. Comparison of tumor-type specific mRNA expression profiles has further provided insight into the biologic differences of these distinct tumor types,17,18,19 and 20 and some candidate biomarkers are being used in diagnostic practice (see below).
Table 7-1 Frequencies of Selected Mutations and Microsatellite Instability (MSI) Across Different Histologic Types | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
LOW-GRADE ENDOMETRIAL CARCINOMA
Low-grade (grade 1 or 2) endometrial carcinomas of endometrioid or mucinous type are associated with low stage at diagnosis and usually a rather favorable outcome. Because pure mucinous carcinomas are rare, the following discussion is restricted to low-grade endometrioid carcinomas irrespective of morphologic variant.
Molecular Pathways
PTEN/PI3K Pathway
Mutations in the tumor suppressor gene PTEN occur in the majority of endometrioid carcinomas with a frequency of 83% in a selected series of endometrioid carcinomas that were associated with a precursor lesion such as endometrial intraepithelial neoplasia.21 PTEN is a dual specific phosphatase that is able to dephosphorylate both proteins and phospholipids. The main substrate of PTEN is phosphatidylinositol-3,4,5-triphosphate (PIP3). PTEN antagonizes the action of phosphatidylinositol 3-kinase (PI3K) on PIP3 with inhibitory effects on the PI3K/AKT signaling pathway. Loss of PTEN function accelerates signaling through the PI3K/AKT pathway. Activation of this pathway results in prevention of apoptosis, in addition to many other cellular effects (Fig. 7-1).22 The PI3K/AKT pathway acts predominantly via the downstream TSC1/TSC2 and rapamycin-sensitive mammalian target of rapamycin (mTOR)
complex. The mTOR complex is a major regulator of ribosome and therefore protein synthesis. External activation of the PI3K pathway can occur via insulin and other growth factors by binding through their receptors. Epidemiologic studies have linked endometrioid carcinomas to individuals with obesity. Obesity is caused by numerous lifestyle factors including high-fat diet and hyperinsulinemia.
complex. The mTOR complex is a major regulator of ribosome and therefore protein synthesis. External activation of the PI3K pathway can occur via insulin and other growth factors by binding through their receptors. Epidemiologic studies have linked endometrioid carcinomas to individuals with obesity. Obesity is caused by numerous lifestyle factors including high-fat diet and hyperinsulinemia.
 FIGURE 7-1: Simplified PTEN/PI3K (left) and Wnt/CTNNB1 (right) signaling pathways including example crossover. Prevalence rates of common mutated genes in endometrioid carcinomas are indicated in percentages. |
PTEN mutations result in loss of function due to frameshift mutations such as insertions or deletions (so called indels), which alter the reading frame (frameshift). A shift in reading frame may lead to nonsense or premature stop codons (nonsense mutation). Nonsense mutation theoretically translates into a nonfunctional or truncated protein. Yet such nonfunctional proteins do not occur due to the nonsense-mediated mRNA decay pathway, which degrades mRNAs containing nonsense mutations before they are translated. Some PTEN mutations are missense mutations that result from a point mutation that leads to substitution of an amino acid. The phosphatase catalytic domain that is encoded on exon 5 of the PTEN gene and missense mutations in this exon are important for loss of PTEN gene function that is accompanied by retained PTEN expression (Fig. 7-2).
Hemizygous PTEN mutations are accompanied by additional genetic alterations of the second allele, such as loss of heterozygosity at chromosome 10q23 leading to biallelic PTEN
inactivation and loss of PTEN expression.21 Loss of PTEN expression is seen in approximately half of endometrioid carcinomas. It is possible that some of the PTEN inactivation is hemizygous. In mice, heterozygous PTEN inactivation produces an abnormal endometrial phenotype in mice, with 100% of mice developing hyperplastic lesions and 20% of animals progressing to endometrial carcinoma.23 The significance of this PTEN haploinsufficiency remains to be elucidated for humans.
inactivation and loss of PTEN expression.21 Loss of PTEN expression is seen in approximately half of endometrioid carcinomas. It is possible that some of the PTEN inactivation is hemizygous. In mice, heterozygous PTEN inactivation produces an abnormal endometrial phenotype in mice, with 100% of mice developing hyperplastic lesions and 20% of animals progressing to endometrial carcinoma.23 The significance of this PTEN haploinsufficiency remains to be elucidated for humans.
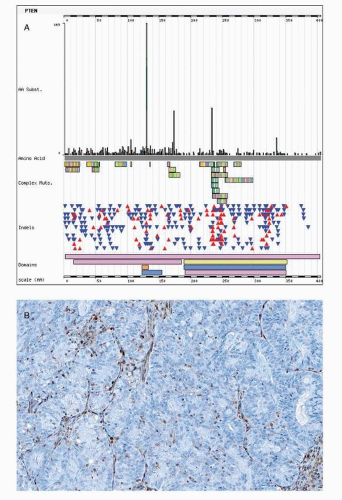 FIGURE 7-2: A: Mutations found in the PTEN gene in a variety of tumor entities and the distribution of these mutations along the protein the gene encodes. The scale represents the 403 amino acids of the PTEN protein. Most mutations are “indels” (i.e., insertion or deletions), which result in a frameshift and a loss of PTEN expression (see part B). Complex mutations result in loss of PTEN expression. There is a small peak of 149 missense mutations (“AA subst”) in exon 5 encoding the phosphatase domain of the gene, which results in a nonfunctional but expressed PTEN protein. (Mutation data obtained from the Sanger Institute Catalogue of Somatic Mutations in Cancer website, http://www.sanger.ac.uk/cosmic; and Bamford S, Dawson E, Forbes S, et al. The COSMIC [Catalogue of Somatic Mutations in Cancer] database and website. Br J Cancer. 2004;91:355-358.) B: PTEN expression by immunohistochemistry can be used as an imperfect surrogate for the PTEN mutational status (i.e., absence of PTEN expression suggests loss of function mutation). |
Inherited germ line mutation of PTEN causes a spectrum of hamartoma syndromes including Cowden syndrome. Besides being associated with benign lesions such as trichilemmomas and thyroid follicular adenomas, Cowden syndrome involves a slightly increased risk for endometrial endometrioid carcinomas, estimated as an increase of 5% to 10% compared to the risk in the general population, with 3% of patients24 indicating a low penetrance of PTEN germ line mutations.25
PTEN mutations have been detected in endometrial hyperplasias, and focal loss of PTEN expression has been observed in approximately half of endometrial hyperplasia, either atypical or nonatypical. Notably, there was no difference in loss of PTEN expression between endometrial hyperplasias that progressed to carcinoma compared with those that did not progress.26 These data suggest that PTEN alterations are common, occur early, and provide a susceptibility background. Loss of PTEN function might be necessary but not sufficient for initiation of endometrioid carcinogenesis. In other words, a few PTEN-negative glands may indicate a low-risk lesion that is likely being shed during subsequent ovulation; however, an expanded field as defined as 1 mm by Mutter (http://www.endometrium.org) may possess the capacity to progress and acquire additional events.
PI3K is another pathway member that is frequently (36%)27 altered by activating mutations. PI3K mutation cluster in exon 9 and exon 20 of the gene PIK3CA encodes the regulatory subunit of class IaA PI3K, p110α. In the study by Oda et al.,27 26% of tumors showed concomitant PTEN and PI3K mutations, indicating a synergistic effect. PI3K mutations, unlike PTEN mutations, were found only rarely in endometrial hyperplasia.28 Histologic grade varied according to location of the PIK3CA mutations. Exon 9 (helical domain) mutations were more often detected in low-grade endometrioid carcinomas compared with high-grade endometrioid carcinomas, which were more often mutated in exon 20 (kinase domain).12 This suggests that PI3K mutations are associated with differentiation of endometrioid carcinomas.
Wnt/CTNNB1 Pathway
Activating mutations in exon 3 of CTNNB1 have been reported in 45% of endometrioid carcinomas.29 Hot spots for mutations are phosphorylation sites of serine-threonine residues coded in exon 3 of CTNNB1 targeted by GSK3 (Fig. 7-3). GSK3 is activated in response to wingless-type mouse mammary tumor virus integration site (Wnt) through the LPR5/6 or frizzled receptors. GSK3 tightly regulates CTNNB1 levels. GSK3 inhibition by lithium increases cytoplasmic CTNNB1 levels.30 Phosphorylation of specific exon 3 residues by GSK3 requires a multiprotein complex including APC and axin, among others, and induces CTNNB1 degradation through ubiquitin-proteasome process. Mutated CTNNB1 escapes this degradation, resulting in increased cytoplasmic level of CTNNB1, nuclear translocation, and enhanced participation in transcriptional regulation through the formation of bipartite complex with the TCF transcription factor. Target genes of Wnt/CTNNB1 signaling include MYC, CCND1, and MMP7.31 The Wnt signaling pathway is involved in pleiotropic cellular functions including differentiation. During embryologic
differentiation, a tight regulation of cellular division and apoptosis is required. With respect to carcinogenesis, the antiapoptotic effects of the CTNNB1 mutation may be the major outcome of aberrant Wnt signaling.
differentiation, a tight regulation of cellular division and apoptosis is required. With respect to carcinogenesis, the antiapoptotic effects of the CTNNB1 mutation may be the major outcome of aberrant Wnt signaling.
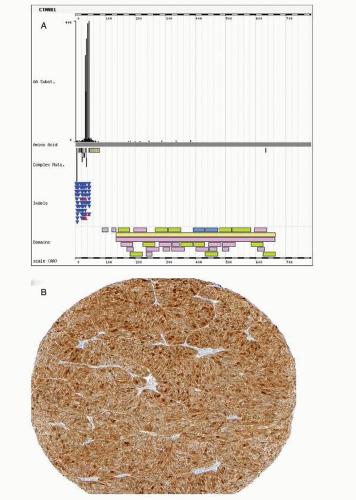 FIGURE 7-3: A: Mutations found in the CTNNB1 gene in a variety of tumor entities and the distribution of these mutations along the protein the gene encodes. CTNNB1 mutations cluster in exon 3 at phosphorylation sites targeted by GSK3. (Mutation data obtained from the Sanger Institute Catalogue of Somatic Mutations in Cancer website, http://www.sanger.ac.uk/cosmic; and Bamford S, Dawson E, Forbes S, et al. The COSMIC [Catalogue of Somatic Mutations in Cancer] database and website. Br J Cancer. 2004;91:355-358.) B: CTNNB1 expression by immunohistochemistry can be used as an imperfect surrogate for the CTNNB1 mutational status (i.e., nuclear and/or cytoplasmic CTNNB1 expression suggests activating CTNNB1 mutation). |
Furthermore, GSK3 provides a cross-link to the tumor suppressor complex TSC1/2. Inhibition of GSK3 will lead to inactivation of TSC1/2, followed by mTOR activation (see Fig. 7-1). This is one example for the complexity of cross talks between the Wnt pathway and other pathways such as the PI3K




Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
