High-Risk Endometrial Carcinoma
Martin Kobel MD
High-risk endometrial carcinomas are a heterogeneous group of tumors that include grade 3 endometrioid, serous, and clear cell carcinomas and other rare types of endometrial carcinoma. The term high risk can be used interchangeably with high grade. High risk is related to the putative outcome of women diagnosed with this disease and is assessed as a risk of tumor-related death of more than 30% to 40% within 5 to 10 years. Each of these tumor types as a group shows high risk, although disease stage-related differences exist.1 There remain significant questions about the natural history of these tumor types. For example, the 5-year survival rate for grade 3 endometrioid carcinomas, all stages, varies from 45% to 77%,1,2 and the rate for serous carcinomas, all stages, ranges from 29% to 55%.1,3 A number of factors could have contributed to the differences in the 5-year survival rate observed in these studies (e.g., extent of surgical staging, use of adjuvant therapy). However, it is also possible that these discordant results with respect to prognosis might be due to differences in accurate classification regarding tumor histologic type of the high-grade endometrial carcinomas. These high-grade tumors particularly represent a clinical challenge because they are uncommon yet account for a disproportionate percentage of tumor-related deaths. Specific studies on molecular alterations in these tumor types have been limited in the past. Currently, these tumors are intensely being studied.
GRADE 3 ENDOMETRIOID CARCINOMAS
The distinction of grade 3 endometrioid carcinomas from low-grade endometrioid tumors as a separate tumor type is based on a distinct phenotype and the presentation at higher stage and high risk of recurrence. However, it is not yet supported by a unique molecular difference, although some differences have been identified. In fact, grade 3 (high-grade) endometrioid carcinomas share the most characteristic abnormalities with grade 1 or 2 (low-grade) endometrioid carcinomas such as similar frequencies of PTEN and PIK3CA mutation (see Table 7-1). Notably, the histologic grade varied according to location of the PIK3CA mutations. Exon 9 (helical domain) mutations were more often detected in low-grade endometrioid carcinomas compared with high-grade endometrioid carcinomas, which more often showed mutations in exon 20 (kinase domain).4
TP53 mutations, a hallmark of serous carcinomas (see below), have been found in 17% of grade 3 endometrioid carcinomas, and TP53 overexpression is detectable in up to 40% of these tumors (Table 8-1).3,5,6,7 and 8 This more than twofold discrepancy again illustrates the need for a more reproducible diagnosis of grade 3 endometrioid carcinomas. The separation of grade 3 endometrioid carcinoma as a unique biologic tumor type may be disputable based
on current knowledge. Yet it is the opinion of the author that understanding of this clinically relevant type will only evolve after adjusting study cohorts for clinical (cohort frequency, stage distribution, outcome) and pathologic criteria. This will allow the cases in different studies to be compared directly and ensure that differences are not related to differences in diagnostic criteria used for type assignment.
on current knowledge. Yet it is the opinion of the author that understanding of this clinically relevant type will only evolve after adjusting study cohorts for clinical (cohort frequency, stage distribution, outcome) and pathologic criteria. This will allow the cases in different studies to be compared directly and ensure that differences are not related to differences in diagnostic criteria used for type assignment.
Table 8-1 Biomarker Expression | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
SEROUS CARCINOMA
Characteristics
Serous carcinomas display severe nuclear atypia, which is the result of chromosomal instability. The hallmark mutation of serous carcinomas is the somatic mutation of TP53 in more than 90% of cases (see Table 7-1). There is a paucity of other recurrent alterations except the high level of genomic instability, which characterizes serous carcinomas and presents a tremendous challenge to identify further relevant alterations. It seems that TP53 mutations are necessary to allow this level of chromosomal rearrangement and to escape the stability control of normal mammalian cells. This assumption is supported by evidence that TP53 mutations are an early event found in precursor lesions of serous carcinomas and from genetically engineered mouse models that reflect serous carcinogenesis. Genetically engineered mouse models developed serous carcinomas exclusively in TP53-mutated background, but TP53 mutation alone was insufficient to generate serous carcinomas. A second oncogenic mutation in a proliferation pathway such as RB1 or MYC was required.9,10 Together, these data suggest that TP53 deficiency is a requirement for serous carcinogenesis (i.e., preventing apoptosis prior to chromosomal instability).
TP53 mutation has been detected in precursor lesions of serous carcinomas.11 The earliest recognizable lesion with evidence of TP53 mutation is the so-called p53 signature.12 p53 signature can be detected by immunohistochemistry as a surrogate for direct sequencing with a minimal requirement of strong nuclear staining in greater than 12 consecutive epithelial cells. p53 signature displays normal morphology without increased proliferation rate, which
delineates it from morphologically recognizable precancers. p53 signatures have been found in the epithelium of 4% of 137 investigated endometrial polyps.13 The significance of p53 signatures remains to be clarified because it is not clear whether these p53 signatures already represent a risk factor or whether p53 signatures represent a lesion with only a minimal risk of progression unless an additional oncogenic hit initiates carcinogenesis.
delineates it from morphologically recognizable precancers. p53 signatures have been found in the epithelium of 4% of 137 investigated endometrial polyps.13 The significance of p53 signatures remains to be clarified because it is not clear whether these p53 signatures already represent a risk factor or whether p53 signatures represent a lesion with only a minimal risk of progression unless an additional oncogenic hit initiates carcinogenesis.
The earliest morphologic recognizable precursor lesions are endometrial glandular dysplasia (EmGD) and endometrial intraepithelial carcinoma (EIC), which are both associated with TP53 mutations. EmGD is defined as glands or surface epithelium displaying a single layer or occasionally two layers of pseudostratification of cells with increased nuclear size (two to three times larger compared with background endometrium but less than four to five times the enlargement seen in EIC).14 TP53 mutations have been detected in 43% of EmGD and 72% of EIC.11 With the assumption that TP53 mutation is a necessity for serous carcinogenesis, the absence of TP53 mutation in a significant portion of precursor lesions needs further elucidation but might be explained by limited sensitivity of direct sequencing, giving the small size of some of those lesions. Although direct sequencing remains the gold standard to detect TP53 mutations, mutational analyses are time consuming and expensive. Therefore, immunohistochemistry has been frequently used as a surrogate marker under the assumption that missense TP53 mutations are associated with nonfunctional protein accumulation, which can be detected by immunohistochemistry with strong staining in more than 50% of tumor cell nuclei.15
A minority of TP53 mutations are nonsense mutations. Nonsense mutations result in a nonsense or premature stop codon, which theoretically translates into a nonfunctional or truncated protein. Yet such nonfunctional proteins do not occur due to the nonsensemediated mRNA decay pathway, which degrades mRNAs containing nonsense mutation before they are translated. As a result, no protein should be present. It remains to be validated whether based on this theoretical framework, TP53 immunohistochemistry can reliably be used as a surrogate marker for the TP53 mutation status. Aberrant TP53 expression would be defined as an overexpression, with more than 50% of tumor cell nuclei expressing the missense mutation, or as completely negative, correlating with the nonsense mutation. In contrast, any expression from single cell to patchy expression would indicate TP53 wild type.
These precursor lesions categorize a morphologic continuum from p53 signature to EmGD to EIC to invasive serous carcinoma (Fig. 8-1). All are characterized by TP53 mutation but differ with respect to the accumulation of additional chromosomal instability reflected by increasing nuclear atypia and proliferation.12 There is a need for further objective criteria that would assign these lesions into meaningful categories for clinical management. For example, EIC is defined as an intraepithelial noninvasive growth pattern but might be already associated with extrauterine disease, although a minority of these cases may actually arise within the distal fallopian tube.16
Compared with other types, serous carcinomas display a high level of chromosomal instability seen as severe nuclear atypia under the microscope or shown by DNA copy number analysis.17,18 As previously mentioned, TP53 mutations do not directly cause chromosomal instability but rather provide the background for the acquisition of such changes by preventing apoptosis. Hence, additional alterations affecting DNA repair, preferentially double-stranded, have been suggested. Ovarian high-grade serous carcinoma is the typical tumor type associated with BRCA1 or BRCA2 mutations.19 Despite some case reports on endometrial serous carcinomas associated with BRCA mutation, the most comprehensive series on 56 cases of endometrial serous carcinomas failed to show any BRCA1 or BRCA2 mutations using tests that would detect approximately 70% of those mutations.20 However,
a recent analysis identified three cases of uterine serous carcinomas associated with BRCA mutation in a population-based series.21
a recent analysis identified three cases of uterine serous carcinomas associated with BRCA mutation in a population-based series.21
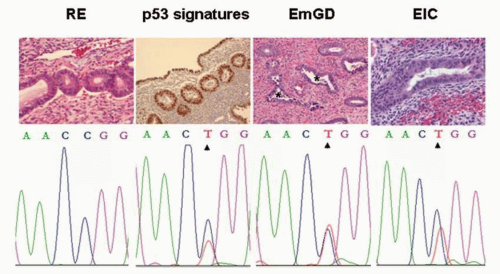 FIGURE 8-1: Proposed sequence of serous carcinogenesis. Top row shows representative images of hematoxylin and eosin staining of resting endometrium (RE), TP53 immunohistochemical staining (p53 signatures), endometrial glandular dysplasia (EmGD), and serous endometrial intraepithelial carcinoma (EIC). Original magnification, ×200. Noticeable degree of nuclear atypia in EmGD (asterisks) clearly exceeds the RE but falls short of EIC. Bottom row shows corresponding DNA sequence analyses that showed identical TP53 gene mutations of exon 7 at codon 248 from CGG to TGG (Arg to Trp), whereas no mutation was found in the corresponding RE sample; results derived from laser capture microdissected samples. These samples were obtained from the same case. Identical mutation was also observed in endometrial serous carcinoma area (not shown) in the same uterus. (With permission from Jarboe EA, Miron A, Carlson JW, et al. Coexisting intraepithelial serous carcinomas of the endometrium and fallopian tube: frequency and potential significance. Int J Gynecol Pathol. 2009;28:308-315.) |
Random chromosomal rearrangements detectable as DNA copy number gains and losses likely activate oncogenes involved in regulating proliferation. Endometrial serous carcinomas are characterized by a high proliferation rate that is already present in its precursor lesions. The Ki-67 labeling index of endometrial serous carcinoma is usually between 50% and 90% of tumor cells. DNA copy number analyses have detected frequent gains at several recurrent chromosomal regions including 8q17,18 containing candidate oncogenes such as MYC.22 Multiple different genetic abnormalities can contribute to a pro-proliferative state by abrogating cell cycle checkpoints, leading to uncontrolled cell cycle progression through G1 and S phases. This further contributes to missegregation of chromosomes during mitosis and fortifying genomic instability. Because a high proliferative rate is characteristic of all serous carcinomas, it is conceivable that all serous carcinomas carry defects in one or more oncogenes regulating proliferation (e.g., cases with amplification of MYC, CCND1, or CCNE1 may be functionally equivalent or synergistic). There has not been a comprehensive survey of all of these alterations in serous carcinoma, and we are left with fragmentary information, typically consisting of data for one or two markers in any given study. We are far from understanding how these
abnormalities co-operate during tumor genesis and how such perturbations in cell cycle control could be used clinically.
abnormalities co-operate during tumor genesis and how such perturbations in cell cycle control could be used clinically.
CDKN2A (p16) seems to be a useful surrogate marker of underlying increased proliferation that characterizes endometrial serous carcinomas. CDKN2A is overexpressed in 90% to 100% of endometrial serous carcinomas using a high cutoff with greater than 80% of tumor cells showing either nuclear or cytoplasmic positive staining.23,24 In contrast to carcinomas of the cervix uteri, endometrial serous carcinomas are not related to human papillomavirus infection. The specific cause for CDKN2A overexpression remains elusive and might represent a futile attempt to stop cell cycle progression, which may be caused by alterations in different cell cycle regulating proteins. This assumption is supported by the concordant overexpression of CDKN2A with CDK2AP2 (p14).
Molecular Prognostic Markers
Related posts:
 Overview of Endometrial Carcinoma
Preneoplastic Conditions of the Endometrium: Endometrial Hyperplasia, the Emergent Concept of Endometrial Intraepithelial Neoplasia, and Others
Endometrial Sarcomas
Staging and Issues Related to Staging of Endometrial Carcinoma
Diagnostic Modalities for Endometrial Carcinoma
Glandular Cell Abnormalities in Pap Tests
Overview of Endometrial Carcinoma
Preneoplastic Conditions of the Endometrium: Endometrial Hyperplasia, the Emergent Concept of Endometrial Intraepithelial Neoplasia, and Others
Endometrial Sarcomas
Staging and Issues Related to Staging of Endometrial Carcinoma
Diagnostic Modalities for Endometrial Carcinoma
Glandular Cell Abnormalities in Pap Tests
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


