Molecular Genetic Aspects of Plasma Cell Disorders
P. Leif Bergsagel
A. Keith Stewart
Stephen J. Russell
Rafael Fonseca
INTRODUCTION
Multiple myeloma (MM) is an age-dependent monoclonal tumor of bone marrow (BM) plasma cells (PCs), often with significant end organ damage that can include lytic bone lesions, anemia, loss of kidney function, immunodeficiency, and amyloid deposits in various tissues.1 It has an estimated incidence of 21,700 in 2012, with 10,710 deaths in the United States.2 Despite recent therapeutic advances, MM continues to be a mostly incurable disease but with a 5-year survival rate reported in the SEER database that has increased from 28% (1987 to 1989) to 43% (2002 to 2008).3 In fact, a subset of younger patients initially treated in 1999 can be identified with a 10-year survival rate of 75%,4 with presumably even better results possible for patients starting treatment today. The incidence is higher in blacks than whites, and in men than women (Fig. 96.1),3 and it is evident from the SEER registry data that although the incidence of MM continues to rise with an annual percentage change of close to 1%, since 1995 the mortality rate has been decreasing at an even faster rate. MM cells are similar to postgerminal center (GC) long-lived PCs, characterized by strong BM dependence, extensive somatic hypermutation (SHM) of immunoglobulin (Ig) genes, and absence of IgM expression in all but 1% of tumors.5 However, MM cells differ from healthy PCs because they retain the potential for a low rate of proliferation (1% to 3% of cycling cells). MM has served as a useful model for understanding the pathogenesis of lymphoid tumors because it is characterized by the presence of a premalignant precursor tumor and defined stages, with researchers able to isolate purified tumor cell populations at all stages.
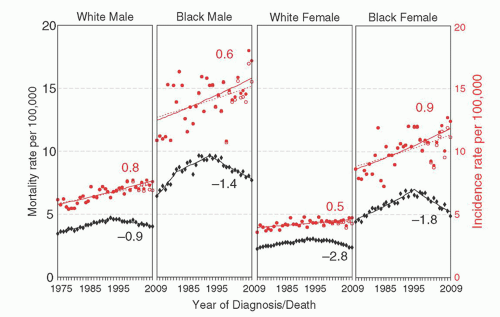 FIGURE 96.1. Differences in US incidence and mortality rates over time, by race and gender. Source is the SEER 9 areas and US mortality files. The most recent annual percentage change for each rate is indicated adjacent to the respective regression line. |
MULTIPLE MYELOMA IS A PLASMA CELL TUMOR OF POSTGERMINAL CENTER B CELLS
Pre-GC B cells can generate short-lived PC that mostly remain in the primary lymphoid tissue (Fig. 96.2). Post-GC B cells can generate plasmablasts (PB) that have successfully completed multiple rounds of SHM and antigen selection, followed by IgH switch recombination, with both B cell-specific DNA modification processes having oncogenic potential.6 These PB typically migrate to the BM, where stromal cells facilitate terminal differentiation into long-lived PC.7 The surface immunophenotype of normal BM PCs is CD38+CD138+ CD19+CD45+CD56–. Although monoclonal gammopathy of undetermined significance (MGUS), SMM, and MM tumor cells also are CD38+CD138+, 90% are CD19–, 99% are CD45– or dim, and 70% are CD56+.8, 9 The reason for this immunophenotypic difference is not known. Possibly there is a normal PC with an MM phenotype, but it is rare, and/or transient, and it has yet to be identified. Alternatively, the change in the phenotype may be a consequence of the transformation process.
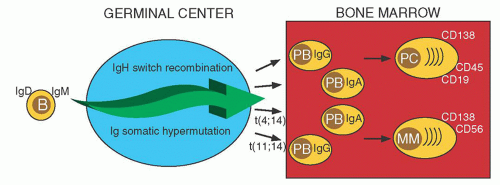 FIGURE 96.2. Normal and malignant plasma cell development. Pre-germinal center (GC) B cells expressing surface immunoglobulin can enter germinal centers where the immunoglobulin genes undergo repeated rounds of somatic hypermutation followed by IgH isotype switch recombination. In multiple myeloma (MM), IgH translocations occur as a result of errors in these two physiologic DNA modification processes (10% and 90%, respectively). Post-GC B cells can generate plasmablast that homes to the bone marrow where stromal cells facilitate differentiation into long-lived PC. Normal plasma cell (PC) express surface CD138, CD19, and CD45; whereas MM cells express CD138, only 10% express CD19, 99% are CD45– or dim, and 70% express CD56. Ig, immunoglobulin; PB, plasmablasts. |
MULTIPLE MYELOMA IS ALMOST ALWAYS PRECEDED BY AN ASYMPTOMATIC MONOCLONAL GAMMOPATHY
An age-dependent premalignant tumor called monoclonal gammopathy of undetermined significance (MGUS)10, 11 is present in about 4% of individuals over the age of 5012, 13 and precedes almost every case of MM. A PC tumor must contain at least a billion cells to produce enough monoclonal Ig (M-Ig) or M-IgL (in the 15% of tumors that produce only IgL) to be detected by serum or urine electrophoresis or a serum-free IgL assay. It can be subclassified as lymphoid (15%) or PC (85%) MGUS, which can progress sporadically at average rates of 1% per year to chronic lymphocytic leukemia/lymphoma/lymphoplasmacytoma/Waldenstrom’s macroglobulinemia and MM, respectively.14 Lymphoid MGUS and PC MGUS can be distinguished by morphology, although clinically the distinction is primarily based on the type of M-Ig detected in serum or urine: mostly IgM for lymphoid MGUS and mostly non-IgM (including Ig light chain only) for PC MGUS. MGUS is distinguished from MM by having a M-Ig of <30 g/L, with no more than 10% of BM mononuclear cells being tumor cells, and no end organ damage or other symptoms. In primary amyloidosis (AL) there is a monoclonal gammopathy which is symptomatic because of pathologic deposits of intact or portions of M-Ig in various tissues.15 Unfortunately, the pathologic deposits can lead to organ failure, so that the median survival of patients with AL is only 3 years, even with a very small number of premalignant tumor cells.16 Progression of MGUS to smoldering MM and symptomatic MM is associated with an expanding BM tumor mass and increasingly severe organ impairment or symptoms.1 Despite BM dependence, sometimes the tumor extends to extramedullary locations, such as spleen, liver, and extracellular spaces. Extramedullary MM (EMM) typically is a more aggressive tumor that sometimes is associated with secondary or primary plasma cell leukemia (PCL), depending on whether or not preceding intramedullary MM was recognized.
A SELECTION FOR GENETIC MUTATIONS THAT ALLOW INDEPENDENCE FROM THE BONE MARROW MICROENVIRONMENT
In considering the diverse array of mutations in MM, it is helpful to place their pathways in the context of the biology of normal BM-localized PCs. MGUS and MM cells, similar to long-lived PC, are dependent on the BM microenvironment, which includes the extracellular matrix and many kinds of cells, e.g., stromal cells, osteoclasts, osteoblasts, immune cells (T lymphocytes, dendritic cells), other hematopoietic cells, and their precursors, vascular endothelial cells.5, 19, 20 These cells mediate reciprocal positive and negative interactions by a variety of adhesion molecules, cytokines, and receptors. Additional stimuli such as hypoxia result in activation of HIF1-α and secretion of VEGF.21 For MM, there are several biologic phenomena that are affected by these tumor-host interactions, including differentiation, and proliferation of tumor cells (most notably IL-6, IGF-1, APRIL); homing to BM; spread to secondary BM sites by the blood stream; generation of many paracrine factors that are involved in survival; angiogenesis; osteoclastogenesis; inhibition of osteogenesis; enhanced resistance to chemotherapeutic agents; humoral and cellular immunodeficiency; and anemia. Many of these tumor-host interactions (e.g., homing, differentiation, and survival) appear to be qualitatively similar for PC and MM tumor cells, whereas the altered composition of the BM microenvironment represents a pathologic response to the MGUS and MM tumor cells. Several therapies (IMiDs, proteasome inhibitors) might target not only the tumor cell but also its interaction with the BM microenvironment. Several of the secondary genetic events associated with the progression of MM discussed below constitutively activate the pathways implicated downstream of the interaction of the MM cell with the BM microenvironment.22, 23
Seven Primary IgH Translocations Are an Early Oncogenic Event in Monoclonal Gammopathy of Undetermined Significance and Multiple Myeloma
Like other B cell tumors, translocations involving the IgH locus (14q32) or one of the IgL loci (κ, 2p12, orλ, 22q11) are common in MM. It seems likely that these events are mediated by errors in IgH switch recombination, or SHM, since the breakpoints are usually located near or within IgH switch regions, but sometimes near VDJ sequences.24 Since there is no evidence that IgH switch recombination or SHM mechanisms are active in normal PC or PC tumors, it is presumed that these translocations usually represent primary—perhaps initiating—oncogenic events as normal B cells pass through GCs. The translocations result in dysregulated or increased expression of an oncogene that is positioned near one or more of the strong Ig enhancers. However, translocations involving an IgH switch region uniquely dissociate the intronic from one or both 3′ IgH enhancers, so that an oncogene might be juxtaposed to an IgH enhancer on either or both of the derivative chromosomes, as first demonstrated for FGFR3 on der(14) and
MMSET on der(4) in MM. Rarely, tumors can have translocations involving two of the primary translocation groups, suggesting that there can be some complementation.18
MMSET on der(4) in MM. Rarely, tumors can have translocations involving two of the primary translocation groups, suggesting that there can be some complementation.18
These IgH translocations are efficiently detected by fluorescent in situ hybridization analyses. Large studies from several groups show that the prevalence of IgH translocations increases with disease stage: about 50% in MGUS or SMM, 55% to 70% for intramedullary MM, 85% in PCL, and >90% in HMCL.25, 26 Limited studies indicate that IgL translocations are present in about 10% of MGUS/SMM tumors, and about 15% to 20% of intramedullary MM tumors and HMCL.18 Translocations involving an IgK locus are rare, occurring in only 1% to 2% of MM tumors and HMCL.18
There are three recurrent primary IgH translocation groups, with the chromosomal sites, target oncogenes, and approximate prevalence in MM (˜40% prevalence for all three groups) as follows: CYCLIN D (11q13-CYCLIN D1-15%; 12p13-CYCLIN D2– < 1%; 6p25-CYCLIN D3-2%) MAF (16q23-MAF-5%; 20q12-MAFB-2%; 8q24.3-MAFA-< 1%; MMSET/(FGFR3)-4p16-(MMSET in all but also FGFR3 in 80% of these tumors)-15%.
Chromosome Content Is Associated with at Least Two Different Oncogenic Pathways
There is a consensus that chromosome content reflects at least two pathways of pathogenesis. Nearly half of MGUS and MM tumors are hyperdiploid (HRD), with 48 to 75 (mostly 49 to 56) chromosomes, usually with extra copies of three or more specific chromosomes (3,5,7,9,11,15,19,21). Nonhyperdiploid (NHRD) tumors have <48 and/or >75 chromosomes. Strikingly, HRD tumors rarely (˜10%) have a primary IgH translocation, whereas NHRD tumors usually (˜70%) have an IgH translocation.27 Tumors with a t(11;14) may represent a distinct category of NHRD tumors as they often are diploid or pseudodiploid. Curiously, EMM tumors and HMCLs nearly always have a NHRD genotype, suggesting that HRD tumors are more stromal cell dependent than NHRD tumors. Although it has been proposed that NHRD and HRD tumors represent different pathways of pathogenesis, the timing, mechanism, and molecular consequences of hyperdiploidy is unknown. Interestingly, in patients with t(4;14) or t(14;16) or t(14;20) or del17p, the presence of one or more trisomies is associated with a substantially better prognosis than with the absence of trisomies. This suggests that the phenotype associated with trisomies may be dominant.28
Universal Cyclin D Dysregulation
Almost all cases of PC neoplasms starting from the MGUS stage express 1 or more of the CYCLIN D genes in an aberrant fashion, despite a low proliferation index.29 Therefore, it has been proposed that dysregulation of a CYCLIN D gene provides a unifying, early oncogenic event in MGUS and MM. MGUS and MM appear closer to normal, nonproliferating PCs than to normal proliferating PB, for which 30% or more of the cells can be in S phase; yet the expression level of CYCLIN D1, CYCLIN D2, or CYCLIN D3 mRNA in MM and MGUS is distinctly higher than in normal PCs. This can be due to direct dysregulation in MM tumors with a CYCLIN D gene translocation or indirectly in tumors with a translocation of MAF, encoding a transcription factor that markedly upregulates CYCLIN D2. Although MMSET/FGFR3 tumors express moderately high levels of CYCLIN D2, the cause of increased CYCLIN D2 expression remains unknown. While normal BM PC express little or no detectable CYCLIN D1, the majority of HRD tumors express CYCLIN D1 biallelically, whereas most other tumors express increased levels of CYCLIN D2 compared to normal BM PC, both by unknown mechanisms. Only a few percent of MM tumors do not express increased levels of a CYCLIN D gene compared to normal PC, but many of these tumors appear to represent samples that are substantially contaminated by normal cells and another large fraction of these tumors often have inactivated RB1, the inhibitor downstream of CYCLIN D, eliminating the necessity of overexpressing a CYCLIN D gene.
Primary IgH Translocations
It is thought that CYCLIN D translocations only dysregulate expression of a CYCLIN D gene. By contrast, MAF translocations dysregulate expression of a MAF transcription factor that causes increased expression of many genes, including CYCLIN D2 and adhesion molecules that are thought to enhance the ability of the tumor cell to interact with the BM microenvironment.29, 30 The contributions of the two genes dysregulated by t(4;14) remain controversial. MMSET is a chromatin-remodeling factor that is overexpressed in all tumors with a t(4;14), whereas about 20% of tumors lack der(14) and FGFR3 expression. The rare acquisition of FGFR3 activating mutations during progression confirms a role for FGFR3 in MM pathogenesis. Although an activated mutant FGFR3 can be oncogenic, it recently was shown that wild-type FGFR3 (as is found in most t[4;14]) can contribute to B cell oncogenesis.31 It remains to be determined if FGFR3 is critical early in pathogenesis but becomes dispensable during progression of t(4;14) MM. Preclinical studies suggest that tyrosine kinase inhibitors are active only against t(4;14) HMCL with activating mutations of FGFR3, whereas anti-FGFR3 monoclonal antibodies that inhibit FGFR3 signaling but also elicit antibody-dependent cell-mediated cyotoxicity are active against HMCLs expressing wild-type FGFR3.32, 33 Despite an apparently indispensable role in t(4;14) MM, it remains to be determined how MMSET, which sometimes has amino-terminal truncations caused by the translocation, contributes to MM pathogenesis. There are some clues. It is a histone methyltransferase for H3K36me2, and when overexpressed it results in a global increase in H3K36 methylation, and a decrease in H3K27 methylation, which might explain some of the many changes in gene expression associated with t(4;14) tumors.29, 34, 35, 36 In addition, it recently has been determined that MMSET has a role in DNA repair (Fig. 96.3). Following DNA damage MMSET is phosphorylated on Ser102 by ATM and is recruited to sites of double strand breaks (DSBs), where it results in methylation of H4K20, which is required for recruitment of p53 binding protein (53BP1). 53BP1 is required for p53 accumulation, G2/M checkpoint arrest, and the intra-S-phase checkpoint in response to ionizing radiation. Approximately half of the translocation breakpoints in t(4;14) MM result in a truncated MMSET that lacks Ser102 and cannot be recruited to DSBs, resulting in a failure to recruit 53BP1 and a loss of the normal DNA damage response pathway. It is not known whether this biologic difference results in a different clinical outcome for t(4;14) MM patients with a truncated versus full-length MMSET.37 Importantly, loss of MMSET expression alters adhesion, suppresses growth, and results in apoptosis of HMCLs, suggesting that it is an attractive therapeutic target.35
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


