Molecular diagnostics in cancer
Roshni D. Kalachand, MBBCh, MD  Bryan T. Hennessy, MD
Bryan T. Hennessy, MD  Robert C. Bast Jr. MD
Robert C. Bast Jr. MD  Gordon B. Mills, MD, PhD
Gordon B. Mills, MD, PhD
Overview
Molecular diagnostics refers to the use of molecular alterations that are associated with cancers to facilitate detection, diagnosis, monitoring, and/or treatment. Molecular biomarkers have often been studied in cancer tissue but potentially can be assayed in readily available patient samples (saliva, sputum, blood, urine, and feces), thereby minimizing the need for invasive biopsies. Traditional blood biomarkers—CEA (carcinoembryonic antigen), PSA (prostate specific antigen), hCG (human chorionic gonadotropin), AFP (alpha–fetoprotein), CA125 and CA15–3—have been used to monitor response to treatment and to detect disease recurrence. Their clinical utility often depends on the availability of effective treatment for residual or recurrent disease. Early detection requires biomarkers with high sensitivity to detect preclinical and ideally premetastatic disease, as well as high specificity to permit efficient, cost–effective screening. Two–stage strategies are often most promising where rising biomarkers trigger imaging or results of imaging are combined with biomarkers to improve positive predictive value. Cancer–specific genomic aberrations have been identified which guide therapy and predict outcomes in subgroups of patients. Trastuzumab has dramatically altered the outcomes of patients with HER2 amplified breast cancers, as have EGFR (epidermal growth factor receptor) inhibitors in the treatment of metastatic EGFR mutation positive non–small–cell lung cancer. As single driver gene aberrations frequently are not sufficient to predict therapeutic responses, gene signatures and multimarker panels incorporating DNA, RNA, and/or protein aberrations are being evaluated as potential effective biomarkers. A panel of multiple biomarkers (OncoType Dx) has proven useful in predicting the need for chemotherapy in addition to hormonal therapy in hormone receptor–positive breast cancer. As targeted therapies are becoming a reality, so is the predictable emergence of resistance to these therapies, often occurring through gene amplification, secondary mutations, or reactivation of signaling mechanisms downstream from the targeted molecular aberration. The search for molecular biomarkers, the so–called companion diagnostics, predictive of first–line therapy resistance and second–line therapy response, is now inherent to targeted drug development. However, these emerging integrative technologies have yet to benefit the majority of patients with cancer. Interpretation of the extensive number of aberrations identified by high–throughput technologies, availability of adequate high–quality tissue, cost, and clinical validation remain significant challenges to developing and implementing effective biomarkers. Strategic use of bioinformatics, international collaboration, development of prospectively collected clinically annotated biobanks containing fresh frozen tissue, and clinical validation in large prospective data sets key to bringing useful molecular biomarkers into clinical practice.
Introduction
Molecular diagnostics involves the use of molecular biomarkers (Box 1) to detect, diagnose, or monitor cancer, as well as to estimate patient prognosis or to predict therapeutic interventions likely to benefit the patient. Ultimately, the development of molecular diagnostics is expected to facilitate the individualization of cancer treatment with the goal of maximizing treatment benefit for individual cancer patients, while minimizing toxicity. Important progress has already been made in developing and applying molecular diagnostics to clinical management. This chapter provides an overview of the current status of molecular diagnostics for cancer. It will also review potential approaches to the integration of biomarker-driven approaches into the future development of individualized cancer therapies, as well as new techniques and likely problems and hurdles that will need to be addressed and overcome.
Box 1 Cancer biomarkers
A cancer biomarker includes any characteristic of tumor cells, stroma, normal tissues, or body fluids that aid in detecting, diagnosing, monitoring, defining prognosis, or predicting response or toxicity from treatment. Thus biomarkers include alterations in DNA, RNA, protein, carbohydrate, lipid, or metabolites as well as biophysical characteristics of tumor cells, the tumor microenvironment, or the host genome and response. Pathologic examination and imaging techniques have provided conventional tumor histologic and staging information that have guided patient management to date. However, these approaches do not consider the full inter- and intratumoral heterogeneity of cancers at the molecular level. Individualization of screening, prevention, and treatment should be substantially improved by considering the profile of multiple molecular biomarkers across different platforms.
Molecular biomarkers for screening and early detection of cancer
Early detection implies the diagnosis of cancer at its earliest stage of development. It is essential that early diagnosis occur at a stage of tumor development where cure can be achieved with currently available therapy. Screening strategies generally require high sensitivity and specificity. Early detection could be facilitated by identifying individuals at high risk for developing specific cancers, decreasing the hurdle of specificity by assessing higher risk individuals.1–4 Screening is a term used for approaches that facilitate detection of tumors at an early curable stage. Effective cancer screening strategies must be cost effective, acceptable to patients and associated with limited morbidity from both the intervention and from false positive results. As screening of the entire population is seldom practicable, guidelines for patient risk assessment are often necessary to appropriately target approaches to prevent and diagnose cancer early. As our understanding of cancer’s molecular heterogeneity advances, novel criteria can be added to the conventional criteria that describe an ideal screening biomarker (Box 2). Thus, as most cancer treatments are effective in only a minority of cancer patients, future useful screening biomarkers could also guide appropriate therapies in individual patients.
Box 2 An optimal screening test
Five critical terms describe the criteria for a screening test: sensitivity, specificity, selectivity, and positive and negative predictive values. Sensitivity is the fraction of cases of the disease that are detected by the test (number of true positives/number of true positives plus false negatives). Specificity is the fraction of cases without cancer that are detected as negative by the test (number of true negatives/number of true negatives plus false positives). Selectivity represents the ability of a screening test to differentiate between different types of cancer. When a test result is a continuous variable, sensitivity and specificity are inversely related and a cutoff for the presence of disease is placed in an arbitrary manner that indicates the clinical consequences of an incorrect result. In most effective screening approaches, a two-stage strategy has proved optimal with a primary diagnostic test followed by a secondary test that optimizes sensitivity and/or specificity beyond that achievable with only a primary test alone (e.g., colposcopy following Pap smear). This approach can help control the consequences of false positive results, invasive approaches, and cost. The prevalence of the disease in a population also affects screening test performance: in low-prevalence settings, even very good tests have poor positive predictive values (the proportion of patients with positive test results who are correctly diagnosed, that is, number of true positives/number of true positives plus false positives). Hence, knowing the prevalence of the disease is a critical prerequisite to interpreting screening test results. Screening for relatively uncommon cancers (e.g., ovarian and pancreatic) requires much greater specificity than screening for more common cancers (e.g., breast or prostate).
Emerging radiologic and endoscopic techniques afford increasingly more sensitive noninvasive procedures for early detection of small tumors. Despite some controversy, low-dose computed tomography appears to detect early lung cancer and decrease lung cancer-related mortality in people at high risk from cigarette smoking, in contrast to routine chest films.5 Similarly, the addition of annual bilateral breast magnetic resonance imaging to annual mammography as an early breast cancer detection strategy for BRCA1/2 gene mutation carriers could decrease the use of bilateral prophylactic mastectomies to reduce the risk of breast cancer in these patients.6, 7 In colon cancer, the ability of colonoscopy to visualize small lesions throughout the entire colon has made this the screening method of choice over double contrast barium enemas. Nonetheless, such tools depend on a tumor’s anatomical features for detection, whereas molecular markers could identify cancer at an early and perhaps premalignant stage, before anatomical detection. Moreover, biomarkers can be detected in bodily fluids (urine, feces, and blood), avoiding potential patient discomfort from endoscopy preparation as well as potentially harmful ionizing radiation from radiologic imaging.
Screening is generally applied to those populations where there is conclusive evidence of an associated survival benefit and, to assure cost effectiveness, where the cancer is a common cause of mortality. As an example, although controversial, most regulatory agencies in the United States recommend annual mammography for women aged 40 years and older.8 Alternatively, risk assessment can be used to stratify select patients to screening or, if the risk is high enough, prevention. This way, the exposure of large numbers of individuals to false positive screening tests and unnecessary biopsies is avoided while maintaining a high cancer detection rate in at-risk patients. Currently, risk assessment is based largely on patient-specific factors, including age, family history, and social factors (e.g., tobacco use), as is the case in lung cancer screening. However, some molecular markers, such as mutations in cancer predisposition genes (e.g., BRCA1, BRCA2, and p53), have demonstrated utility in the identification of at-risk populations.
Molecular biomarkers in current clinical practice for screening, prevention and early detection
Prostate cancer
Prostate-specific antigen (PSA) is normally present in the blood at very low levels ranging between 0 and 4.0 ng/mL. Increased PSA levels can be associated with an underlying prostate cancer. Serum PSA measurement for prostate cancer screening is controversial, despite widespread use.9 Up to 15% of prostate cancers can occur in the absence of an elevated PSA. PSA levels can be elevated due to prostate infection, irritation, benign prostatic hypertrophy, or recent ejaculation. Thus, PSA is not an adequately sensitive or specific marker for prostate cancer screening. PSA screening can lead to a high rate of unnecessary biopsies, over-diagnosis and overtreatment, resulting in morbidity. Both prostatectomy and radiotherapy can be associated with impotence and incontinence. Patients diagnosed with less aggressive (Gleason score < 7) prostate cancer can survive decades with their cancer, often dying from comorbid diseases. PSA screening in men over the age of 50 years can confer up to a 20% decrease in prostate-cancer-specific mortality, although this may or may not translate into a decrease in overall mortality, judging from large multicenter trials.10, 11 In the past, PSA screening has been recommended for men older than 50 years with an expected life expectancy of 10 years or more, following discussion with the patient about screening-associated risks.12 The United States Preventive Health Task Force now recommends against using PSA for screening.13 The American Urological Association (AUA) has more nuanced age-dependent guidelines.14 The AUA recommends against PSA screening for men under age 40. Between age 40 and 54, PSA screening is recommended only for African-American men or men with a positive family history conferring increased risk. For men ages 55–69, the benefits of preventing prostate cancer mortality in one man for every 1000 men screened over a decade must be weighed against the known harms associated with screening and treatment. PSA screening is not recommended for men over 70 years.
Most PSA in the blood is bound to serum proteins. A small amount is not, and is called free PSA, an isoform of which, [-2]proPSA, is highly associated with prostate cancer. The combination of PSA, free PSA, and [-2]proPSA in a single score, the Prostate Health Index (PHI), shows increased sensitivity over PSA screening alone in men with PSA levels between 4 and 10 ng/mL but has not been widely adopted as a prostate cancer screening tool.15
Ovarian cancer
The relatively low prevalence of ovarian cancer—1 : 2500 in the United States—means that strategies for early ovarian cancer detection must have relatively high sensitivity (>75%) for preclinical disease and an extremely high specificity (99.6%) to attain a positive predictive value of at least 10%, that is, 10 operations for each case of ovarian cancer detected. Serum CA125 has received the most attention but lacks the sensitivity or specificity to function as a stand-alone screening test. Two-stage screening strategies promise to be more effective, where increasing levels of serum biomarkers over time prompt transvaginal sonography (TVS) to detect lesions that require laparotomy. With annual determination of CA125 in women at average risk for ovarian cancer, a computer algorithm has been developed to determine deviations from each woman’s own baseline. If CA125 increases significantly, TVS is performed and if imaging is abnormal, laparotomy is undertaken. If the CA125 is unchanged, a woman returns in 1 year and if CA125 is mildly elevated, biomarker levels are obtained in 3 months. On the basis of this strategy, a study of more than 5000 healthy postmenopausal women coordinated by MD Anderson, performed 18 operations to detect 2 borderline and 10 invasive cancers with 75% in stage I or II.16 No more than three operations were required to detect each cancer. A much larger randomized trial involving 200,000 in the United Kingdom is powered to test the impact of this strategy on survival and mortality. Data from the first 2 years of accrual suggest that a two-stage strategy could increase the fraction of disease detected in early stage.17 Using deviations from each woman’s baseline, rather than a single cutoff for CA125 alone, doubled the number of ovarian cancer cases detected.18 Outcome of the study will be reported in the near future and could change practice, if positive. As only 80% of ovarian cancers express CA125, other markers will be required for early detection of all ovarian cancers. The development of technologies that measure multiple serum markers simultaneously, linked to the creation of statistical methods that enhance sensitivity without sacrificing specificity, hold great promise.
Cervical cancer
The establishment of national cervical cancer screening programs involving the detection and treatment of the premalignant stage of cervical intra-epithelial neoplasia (CIN) through cervical Pap smear cytology has reduced incidence and mortality from cervical cancer by 80%. Isolated Pap smears have false negative rates of 20–40%, which are partly overcome by regular screening (every 3 years). The human papilloma virus (HPV), in particular strains 16 and 18, is critical to cervical carcinogenesis. Testing for HPV DNA in cervical smears increases the cervical cancer detection by 30% over cytology-based screening and could lengthen the screening interval to 5 years.19 Incorporation of HPV-based screening into guidelines is being considered. Importantly, however, the Pap smear and HPV DNA function as primary screens that are followed by visual and pathologic evaluation through colposcopy as a secondary screen.
Individuals at elevated risk
Specific guidelines exist for patients with strong family histories of cancer, particularly for carriers of genomic biomarkers of high cancer risk. When the cancer risk associated with the specific (usually hereditary) biomarker is high enough, recommendations focus on prevention rather than early detection. As prevention strategies generally impact quality of life to a greater degree than does screening, the identification of inherited mutations associated with a very high cancer risk necessitates patient education and careful shared decision making. Furthermore, hereditary biomarkers testing has significant implications for family members. Prophylactic surgery and chemoprevention are particularly effective risk reduction techniques that are reserved for people at highest risk. Prophylactic oophorectomy and/or mastectomy has the greatest protective effect for BRCA1/BRCA2 mutation carriers, reducing the risk of ovarian cancer and breast cancer by more than 90%.20, 21 However, some patients prefer mammography, MRI, or other screening approaches to delay surgical intervention either for personal preference or to allow childbearing. There are many factors (e.g., ethics and cost) to consider in discussing options for early detection and prevention with very high-risk patients, and this area of medicine is a rapidly evolving specialty.22–24
Breast and ovarian cancer
In families with a significant history of breast and/or ovarian cancer, key cancer risk biomarkers (usually hereditary mutations) can now be identified in a minority of cases, using clinical prediction models (e.g., BRCAPRO model).25 Such models can be useful in guiding specific molecular tests that further stratify risk and guide subsequent screening or prevention. Inherited mutations in BRCA1 and BRCA2 account for 5–10% and 10–15% of all breast cancers and ovarian cancers, respectively.26, 27 Furthermore, approximately 15% of patients with triple negative breast cancers or with high-grade serous ovarian cancer will carry deleterious germ line mutations in BRCA1/2 in the absence of a significant family history. Moreover, BRCA1/2 mutations guide therapy in ovarian cancer, as outlined in the following section. Approximately 12% of women at high-risk do not carry BRCA1/2 mutations and are estimated to have another cancer predisposing genomic alteration,28 suggesting a need for improved testing approaches that identify additional risk biomarkers. Less well-studied breast cancer susceptibility genes include CHEK2, ATM, RAD51C, BRIP1, PALB2, NBS1, LKB1, PTEN, p53, XRCC1, and STK11, but these are not commonly assessed due to the rarity of inherited mutations.29–32 Recently, these genes have been integrated into multiplex sequencing assays, both providing more information and greater challenges in patient decision making for genes associated with a lower frequency of cancer development.
Colon cancer
Familial adenomatous polyposis (FAP) and hereditary nonpolyposis colon cancer (HNPCC) predispose to early-onset familial/hereditary colon cancer. They are characterized by germ line mutations in the adenomatous polyposis coli (APC) and DNA mismatch repair genes, respectively. In HNPCC, routine tumor molecular screening using antibodies to mismatch repair proteins has superseded clinical risk models to guide screening (e.g., revised Bethesda guidelines). Tumor molecular screening, however, results in excessive genetic testing for HNPCC, as 10–15% of cases represent sporadic disease.33 Combining the clinical risk prediction models with tumor molecular screening, or testing the tumor for molecular markers of sporadic disease (MLH1 promoter methylation and/or BRAF V600E gene mutation) could improve screening for HNPCC while maintaining high detection rates.34 Management guidelines entail surgical cancer prevention (colectomy) with FAP and intensive colonoscopic screening with HNPCC.
In a recent study, fecal hemoglobin detection was combined with fecal DNA tests for mutant K-Ras, aberrant NDRG4 and BMP3 methylation and B-actin.35 When compared to a standard fecal immmunohistochemical test (FIT) in 9989 evaluable participants at average risk, the composite Cologuard® test demonstrated greater sensitivity for detecting colorectal cancer (92.3% vs 73.8%, P = 0.002), greater sensitivity for detecting advanced precancerous lesions (42.4% vs 23.8%, P < 0.001), a higher rate of detection of high-grade dysplastic polyps (69.2% vs 46.2%, P = 0.004), and greater detection of serrated sessile polyps measuring 1 cm or more (42.4% and 5.1%, P < 0.001). Specificity, however, was somewhat lower with DNA testing than with FIT (86.6% vs 94.9%). On the basis of these results, the test was approved in 2014 by the United States FDA for individuals greater than 50 years of age.
Future approaches to early detection and screening
While current approaches to early detection have impacted mortality from certain forms of malignancy such as cervical cancer, low sensitivity, unnecessary use of invasive diagnostic procedures due to false positives, and overdiagnosis/treatment remain significant issues. Moreover, many tumor types, such as ovarian and pancreatic cancer, do not benefit from these approaches. Patients continue to present with advanced disease and thus have poor outcomes. Elevation of circulating tumor biomarkers may require a substantial tumor volume. Autoantibodies could be evoked by small volumes of cancer at an earlier interval. Novel molecular screening techniques, including circulating DNA, RNA, and exosomes, have the potential to complement current protein-based screening markers and revolutionize early cancer detection, while facilitating accurate risk assessment and individual treatment planning.
Current molecular biomarkers for predicting outcomes and therapy responsiveness
Although molecular biomarkers that predict outcomes in cancer patients are useful, greater clinical utility lies in biomarkers that predict benefit from specific cancer therapies. There is considerable overlap between both types, as those biomarkers that predict benefit for particular patients from specific cancer therapies will predict improved outcomes.
Breast cancer
Hormone receptors (HR)
Hormone receptors (HR)-positive breast cancer comprises approximately 70% of all breast cancers and is marked by the expression of estrogen receptor (ER) alpha and/or progesterone receptor (PR). The HR biomarkers identify breast tumors that are sensitive to growth inhibition by antihormonal treatments, including ER partial agonists/antagonists (e.g., tamoxifen), ER downregulators (e.g., fulvestrant), and aromatase inhibitors (e.g., letrozole).36, 37 In clinical practice, HR protein expression is assessed routinely in all breast cancers using immunohistochemistry. Despite 5 years of adjuvant antihormonal therapy, however, a significant fraction of women with early stage HR-positive breast cancer relapse, and the majority of women with metastatic HR-positive breast cancer, develop resistance to antihormonal manipulation. Thus, in the United States, over 25,000 women with HR-positive breast tumors die each year, more annual deaths than are caused by all other types of breast cancer combined.
Multiparameter gene expression profiles
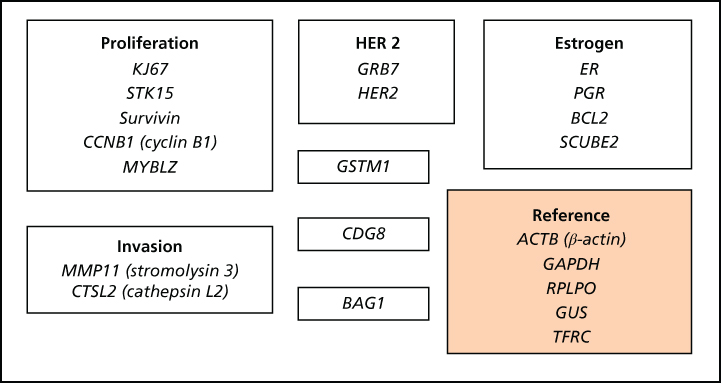
Our ability to predict the likelihood of cure for patients with HR-positive breast cancer after treatment with antihormonal drugs has improved dramatically. Oncotype Dx (Table 1), based on the expression of 21 genes, predicts the benefit for individual node-negative HR-positive breast tumor patients from adjuvant tamoxifen and selects patients for cytotoxic chemotherapy based on features associated with tamoxifen resistance.38 However, despite this approach’s clinical utility, this and similar assays such as PAM50 and Mammoprint do not increase our understanding of antihormone resistance mechanisms in HR-positive breast cancer beyond the known roles of tumor grade, HER2, and HR levels.36, 38, 39 The phosphatidylinositol-3-kinase (PI3K)/AKT/mTOR and mitogen-activated protein kinase (MAPK) pathways are major mediators of the effects of membrane receptor tyrosine kinases (RTKs) such as HER2 and mediate resistance to antihormonal therapies.40 The mTOR inhibitor everolimus improves progression-free survival for approximately 60% of women with HR-positive breast cancers resistant to antihormonal therapies.41 A predictive molecular biomarker of response to this drug is lacking however.
HER2
The oncogene encoding HER2 is amplified and the protein overexpressed in 15–20% of invasive breast cancers. HER2 overexpression dictates an aggressive breast tumor phenotype and poor prognosis.42 Combining trastuzumab (Herceptin), a recombinant humanized monoclonal antibody targeting HER2, with cytotoxic chemotherapy to treat patients with metastatic and early stage HER2 oncogene-amplified breast cancer, has resulted in increased response rates and improved survival.43 Thus, HER2 is a biomarker for breast tumor responsiveness to trastuzumab and other HER2-targeted therapies (e.g., pertuzumab, lapatinib, and TDM1).44 More recently, HER2 has been validated also as a biomarker predictive of response to trastuzumab-based chemotherapy in 15–20% of advanced gastric cancer patients with HER2-driven oncogenesis.45
A significant fraction of HER2-amplified breast and gastric cancers do not respond initially to trastuzumab or acquire resistance to trastuzumab. This could be mediated by a cleaved form of HER2 that does not bind trastuzumab (p95-HER2), by upregulation of other membrane RTKs such as IGF1R or MET or by upregulation of the PI3K/AKT pathway.46–49 The latter can occur through inactivating mutations of PTEN (a negative PI3K/AKT regulator), PTEN loss, or by PIK3CA mutations, the oncogene that encodes the p110α subunit of PI3K.50 Indeed, PI3K pathway activation and mutations in PIK3CA and PTEN are negative prognostic markers in HER2-positive breast cancers treated with anti-HER2 targeted therapies.51
A panel of the American Society of Clinical Oncology (ASCO) thus recommended that HER2 status should be determined for all invasive breast cancers.52 This panel has proposed a testing algorithm that relies on accurate, reproducible assay performance, including newly available types of bright-field in situ hybridization (ISH), and has specified elements to reliably reduce assay variation (e.g., specimen handling, assay exclusion, and reporting criteria). This emphasizes the need for quality control and validation approaches for all predictive markers, both in terms of reliability and clinical utility.
Ovarian cancer: BRCA1/2
Approximately 20% of high-grade serous ovarian cancers have an underlying inherited or somatic BRCA1/2 mutation, rendering them deficient in homologous recombination, a pathway essential for repairing double-stranded DNA breaks.53 These tumors rely on alternative DNA repair mechanisms such as base excision repair, to which the PARP enzyme is key. Inhibitors of PARP exploit vulnerability of BRCA1/2-mutated ovarian cancers to DNA damage. In a randomized phase II trial, the PARP inhibitor olaparib increased progression-free survival by 82% as compared to placebo when used as maintenance therapy in platinum-sensitive recurrent BRCA1/2-mutated ovarian tumors.54 Olaparib has now been approved for use with this indication, with approvals for other PARP inhibitors and other uses expected imminently. Resistance mechanisms include function-restoring BRCA1/2 mutations, loss of 53BP1 (a protein involved in an alternative DNA repair mechanism), and upregulation of the PI3K/AKT/mTOR pathway.55 The combination of olaparib and BKM120, a PI3K inhibitor, has shown activity in early clinical studies of high-grade serous ovarian and triple negative breast cancer.56
Lung cancer
Epidermal growth factor receptor
The reversible EGFR (epidermal growth factor receptor) tyrosine kinase inhibitors (TKIs) gefitinib and erlotinib and irreversible pan-HER TKI afatinib and dacomitinib provide effective therapies in approximately 10–30% of patients with non-small-cell lung cancer, whose tumors harbor activating EGFR mutations, the common ones being located on exon 19 and 21.57–60 Female nonsmokers of Asian origin with adenocarcinoma represent the typical phenotype, although EGFR mutations are also observed in men and former smokers. While first line treatment with dacomitinib in EGFR-mutant lung cancers has been associated with an impressive progression-free survival (18.2 months), and afatinib shows greatest efficacy in exon 19 mutations, the benefit of one TKI over another has yet to be established.
Multiple acquired resistance mechanisms to EGFR inhibitors, including secondary mutation in EGFR (T790M) and MET amplification,61 limit the efficacy of EGFR-targeted therapy. New EGFR TKIs have shown promise in treating or preventing resistance in phase I/II studies and are being evaluated in larger clinical trials,62 with the hope that they will translate into effective therapies for patients with lung cancer.
ALK and ROS-1
Two targetable chromosomal gene rearrangements leading to oncogenic gene fusions guide novel therapies in advanced adenocarcinoma of the lung.63, 64 ALK (anaplastic lymphoma kinase) and ROS-1 gene translocations are present in 4–6% and 1–2% of advanced lung adenocarcinomas, respectively, rendering them markedly sensitive to the ALK, ROS-1, and MET inhibitor, crizotinib. The short period between the identification of these targets and crizotinib’s approval engenders excitement for an evolution away from the prolonged time period traditionally required for drug discovery and implementation. Crizotinib is associated with response rates of over 70% and a median progression-free survival of 11 and 19 months for ALK and ROS-1 rearranged tumors, respectively. Gene overexpression, bypass mechanisms, and secondary mutations commonly cause acquired resistance, though unlike EGFR-mutated non-small-cell lung cancer, multiple mutation types are observed in a single tumor at disease progression. Strategies to overcome acquired resistance involving second-generation ALK inhibitors such as ceritinib show promise in early clinical trials.65
On the basis of these results, routine molecular testing is recommended for EGFR and ALK in all advanced stage lung adenocarcinomas (or lung tumors with an adenocarcinoma component), regardless of clinical characteristics. Tumors that do not contain an adenocarcinoma component perhaps due to a sampling error but are suspected as such due to clinical characteristics (young, never smoker) may also be tested.66
Colon cancer
Microsatellite instability
Current guidelines recommend adjuvant 5-fluorouracil-based chemotherapy in stage II colon cancer in “high-risk” patients, defined by the presence of high-grade tumors with lymphovascular space invasion, evidence of tumor perforation, and/or less than 12 nodes extracted at surgery. Approximately 11% of stage II colon cancers have a deficiency in mismatch DNA repair, manifested as microsatellite instability.67 Such tumors have good outcomes in the absence of adjuvant chemotherapy. Therefore, guidelines advise routine immunohistochemical testing of stage II colon cancers for mismatch repair proteins, the absence of which obviates the need for adjuvant chemotherapy.
KRAS, BRAF, and NRAS
KRAS is a proto-oncogene downstream of EGFR that initiates signaling through the Ras-Raf-MAPK pathway. Approximately 60% of metastatic colorectal cancers have the favorable wild type KRAS profile, predicting sensitivity to the anti-EGFR monoclonal antibodies, cetuximab and panitumumab. KRAS mutations, found mainly at codons 12 and 13 of exon 2, predict resistance to these therapies, as do the rarer NRAS mutations.68–71 Downstream BRAF mutations occur in about 5–9% of cases and correlate with poor prognosis, although their reported lack of benefit from EGFR inhibitors remains uncertain.
Gastrointestinal stromal tumors (GIST): KIT and PDGFRA
GIST (gastrointestinal stromal tumor) is associated with primary activating mutations in the KIT (80% of GISTs) or platelet-derived growth factor receptor A (PDGFRA; 5–10% of GISTs) genes that result in constitutive RTK activation.72 Imatinib (Gleevec), which inhibits both KIT and PDGFRA, results in clinical benefit in approximately 85% of patients with unresectable or metastatic disease, with a median progression-free survival of 20–24 months, although patients with exon 11 mutations fare better than exon 9 mutations. In the latter case, a higher dose of imatinib confers a greater benefit. The mechanisms of acquired resistance to imatinib are heterogeneous, involving mostly the emergence of secondary mutations in KIT exons 13, 14, or 17. In patients with imatinib-resistant GIST, novel kinase inhibitors, such as sunitinib, nilotinib, dasatinib, and regorafenib, can inhibit the mutant protein’s function and restore antitumor activity.
Melanoma
BRAF
About half of all melanomas carry the BRAF V600E gene mutation, leading to constitutive downstream activation of the MAPK pathway engendering sensitivity to BRAF inhibitors (e.g., vemurafenib and dabrafenib).73, 74 Clinically, the duration of response to these drugs is short, with progression occurring at a median of 6–7 months. Novel approaches to overcome resistance resulting from compensatory overactivation of the MAPK pathway involve the upfront combination of BRAF and MEK inhibitors (such as trametinib), which increases the efficacy and decreases the toxicity of therapy.75
NRAS
MEK inhibitors will likely play a significant role in treating NRAS-mutated melanoma, which involves approximately 15–20% of all melanomas and dictates a particularly aggressive biology. Interestingly, MEK inhibitors may demonstrate activity in combination with immune therapies.76
KIT
A small number (3%) of BRAF/NRAS-negative melanomas carry KIT proto-oncogene mutations, particularly in mucosal and acral melanomas. Targeted therapies with imatinib or sunitinib are viable therapeutic options that can provide durable responses, with evidence of nilotinib activity at progression post imatinib therapy.77, 78
Chronic myeloid leukemia (CML)
Bcr-Abl translocation
Most chronic myeloid leukemia (CML) cases are driven by constitutive activation of the Abl kinase as a result of the breakpoint cluster region (Bcr) to the Abl kinase translocation (Philadelphia chromosome). Imatinib targeted therapy, which inhibits the kinase activity of Abl, has markedly improved the outlook for patients with CML. However, imatinib resistance can emerge despite initial benefit. Furthermore, efficacy of imatinib is limited in advanced “blast-crisis” CML.79 Resistance is due predominantly to novel mutations in the Abl kinase domain, interfering with drug binding. Thus, new Abl kinase inhibitors (AKIs) were developed, among which nilotinib, dasatinib, and bosutinib gained regulatory approval. Unfortunately, all available AKIs exhibit inactivity to certain kinase domain mutations, the cross-resistant Bcr-Abl (T315I) mutant being the most common. The third-generation AKI ponatinib demonstrates clinical benefit in Bcr-Abl (T315l) mutant CML and some other multidrug-resistant mutations. However, cardiovascular toxicity concerns, potentially related to the choice of a high-drug dose during clinical development, precluded regulatory approval. Trials investigating dose-optimized ponatinib are underway.80
Lymphoma
CD20 and other biomarkers
Biomarker-targeted therapeuties have impacted significantly the treatment of non-Hodgkin lymphoma (NHL).81, 82 The development of the chimeric anti-CD20 antibody rituximab heralded a new era in NHL treatment approaches. Rituximab is now standard monotherapy for front-line treatment of follicular lymphoma and is used in conjunction with chemotherapy for other CD20-positive B-cell lymphomas. The subsequent development and approval of radio-immunoconjugates of rituximab (90Y-ibritumomab tiuxetan and 131I-tositumomab) has improved outcomes further.
Clinical evaluation of antibodies has been based largely on knowledge of antigen expression on the lymphoma cells’ surface, leading to the development of antibodies against CD22 (e.g., unconjugated epratuzumab), CD80 (galiximab), CD52 (alemtuzumab), CD2 (siplizumab), CD40 (SGN-40), and CD30.83 The anti-CD30 antibody brentuximab has been conjugated with a drug, vedotin, to target cytotoxic drug delivery to CD30-positive Hodgkin’s or anaplastic large cell lymphoma, producing significant clinical benefit with acceptable toxicity. It was approved for use recently in relapsed Hodgkin’s lymphoma following autologous stem cell transplant and relapsed anaplastic large cell lymphoma. Currently, it is being investigated for use upfront with chemotherapy.
Oropharyngeal cancer (OPC)
Human papilloma virus
The epidemiology of oropharyngeal cancer (OPC) has changed over the last two decades with studies reporting the presence of HPV p16 subtype in 60–70% of all OPC in western countries. Molecular tumor testing is easily done using p16 immunohistochemistry, which has a high concordance with HPV FISH. Retrospective analyses of large clinical trials demonstrate superior response rates and survival with chemoradiotherapy in HPV-related OPCs, as compared to HPV-negative OPCs.84 Therefore, HPV is an important stratification tool for future therapeutic investigations in OPCs and may dictate a different therapeutic approach. Suggestions that HPV-associated OPCs may benefit from less intensive chemoradiotherapy require further evaluation.
Molecular biomarkers for monitoring of cancer
A number of circulating biomarkers are used for monitoring of cancer response to therapy and/or for the early detection of recurrent disease in cancer patients.
Alpha-fetoprotein (AFP) and human chorionic gonadotropin (hCG)
Many germ cell tumors [most male testicular cancers, gestational trophoblastic disease (choriocarcinoma), and rare ovarian cancers] produce circulating tumor markers [AFP (alpha-fetoprotein), hCG (human chorionic gonadotropin), lactate dehydrogenase (LDH)].67 These biomarkers are useful in diagnosing, staging, monitoring therapeutic response, and detecting early tumor recurrence. As recurrent germ cell tumors can be cured with cytotoxic chemotherapy, particularly when the recurrence is detected early, increasing tumor marker levels during patient follow-up are an indication for initiating salvage therapy, despite absence of evident disease. The markers’ half-lives must be considered when evaluating therapeutic responses.
AFP is a glycoprotein normally produced by the fetal yolk sac, liver, and gastrointestinal tract but not by normal adult tissues. It is re-expressed in germ cell tumors, including yolk sac tumors and embryonal carcinomas. hCG is a glycoprotein produced by syncytiotrophoblastic cells consisting of two subunits, α and β. The α-subunit is common to three pituitary trophic hormones: FSH, LH, and TSH; the β-subunit makes hCG enzymatically and immunologically distinct. Assays measure only the β subunit (β-hCG). In males, it is highly specific for testicular cancer, in particular choriocarcinoma cells and 5–10% of pure seminomas. LDH reflects “tumor burden,” growth rate, and cellular proliferation and has independent prognostic significance. LDH is increased in about 80% of advanced seminomas and about 60% of advanced nonseminomatous germ cell tumors. The LDH isoenzyme 1 seems to be more specific and sensitive for germ cell tumors than isoenzymes 2–5.85
CA125
CA125 is a mucinous transmembrane glycoprotein product of the MUC16 gene that ranges up to 5 mD. CA125 is best known as an ovarian cancer marker, though elevations in endometrial, fallopian tube, lung, breast, and gastrointestinal cancers, as well as relatively benign conditions including endometriosis, are observed. CA125, when used alone on a single occasion, is not sensitive or specific enough for ovarian cancer screening.86 Moreover, 20% of ovarian cancers do not produce elevated CA125 levels. However, serum CA125 is very useful for following treatment response, for predicting post-therapy prognosis, and for detecting recurrence in women with ovarian cancer. During first-line platinum-based ovarian cancer chemotherapy, CA125 levels should be followed regularly (e.g., every 3 weeks). The CA125 level at nadir, 3-month normalization of CA125 and CA125 half-life are strong predictors of progression-free and overall survival times.87–89 The failure of serum CA125 normalization after initial treatment with surgery and platinum-based chemotherapy is a particularly ominous indication of poor prognosis in women with ovarian cancer.
In women in clinical remission following previously treated ovarian cancer, the National Comprehensive Cancer Network (NCCN) (www.nccn.org) recommends evaluation of serum CA125 level at each follow-up visit if the CA125 level was elevated at the initial diagnosis. After a documentated CA125 elevation, the median time to clinical disease relapse is over 4 months, although increases within the normal range can produce much longer lead times. One large, randomized study in the United Kingdom concluded a lack of overall survival benefit when women with recurrent disease were treated at the time of CA125 elevation, although only 25% of women on the control and experimental arms were treated promptly with optimal combination chemotherapy.90 At a minimum, earlier recurrence detection with CA125 provides time to receive known and novel agents, given the relatively short interval between symptomatic recurrence and death. With such controversy, however, using CA125 to monitor recurrence should be discussed with each patient.
CA15-3 and CA27.29
CA15-3 and CA 27.29 are well-characterized assays that detect circulating MUC1 antigen in peripheral blood.91 Several studies support the prognostic relevance of this circulating marker in early stage breast cancer though monitoring MUC1-based serum markers has demonstrated utility in making treatment decisions.92, 93 ASCO regards available data as insufficient to recommend using CA15-3 or CA 27.29 for breast cancer screening, diagnosis, staging, or for monitoring patients for recurrence, as conclusive evidence that early detection of recurrence improves survival is lacking.91 Although present data cannot recommend the use of CA15-3 or CA 27.29 alone for monitoring treatment response, rising CA15-3 or CA27.29 may be used to indicate treatment failure in the absence of readily measurable disease. However, caution should be used when interpreting a rising CA27.29 or CA15-3 level during the first 4–6 weeks of a new therapy, as spurious early rises may occur. These recommendations apply also when monitoring metastatic colon cancer with CEA (carcinoembryonic antigen).
CA19-9
Carbohydrate antigen 19-9 is elevated primarily in the serum of patients with gastrointestinal tract carcinomas. The greatest utility of serum CA19-9 is in monitoring treatment response in pancreatic cancer. For patients with locally advanced or metastatic pancreatic cancer undergoing active therapy, ASCO recommends measurement of serum CA19-9 levels every 1–3 months.94 Serial CA19-9 elevations suggest progressive disease in the face of treatment, but confirmatory studies (e.g., CT scanning) should be sought before a therapy change is initiated.
Carcinoembryonic antigen (CEA)
CEA is a cell adhesion glycoprotein.95 It is produced during fetal development and is not usually present in healthy adults’ blood, although levels are raised in heavy smokers. Serum CEA may be elevated in patients with colorectal, gastric, pancreatic, lung, breast, and medullary thyroid carcinomas. ASCO has developed clinical practice guidelines to monitor serum CEA levels in patients with colorectal cancer, in whom serum CEA should be ordered preoperatively, if it would assist in staging and surgical planning. Postoperative CEA levels should be performed every 3 months, for patients with stages II and III colorectal cancer, for at least 3 years if the patient is a potential candidate for surgery (e.g., liver resection) or chemotherapy for metastatic disease. CEA is also the marker of choice for monitoring systemic therapy response in metastatic colorectal cancer.
Prostate specific antigen
Measuring serum PSA is important in men with an established diagnosis of prostate cancer. The rate of PSA rise can predict prostate cancer prognosis. Men with prostate cancer whose PSA level increased by more than 2.0 ng/mL during the year before the diagnosis of prostate cancer have a higher risk of death from prostate cancer after radical prostatectomy. PSA level along with clinical stage and Gleason tumor grade are components of most nomograms and predictive models used for prostate cancer risk assessment.96 Further, serum PSA provides an indicator of disease response to treatment.
For prostate cancer patients treated initially with curative intent, serum PSA level should be assessed every 6–12 months for 5 years and annually thereafter. A rising PSA level indicates biochemical failure and often precedes a clinically detectable recurrence by several years. As biochemical failure may represent an isolated local recurrence, identifying those patients is important as they may be candidates for salvage therapy.
Novel molecular biomarkers and platforms for their detection
Malignant tumors are characterized by multiple molecular anomalies responsible for the behavior of individual cancers. The driving aberrations can be in the germ line genome or may occur in a somatic or acquired manner in the cancer genome and/or proteome. Novel cancer biomarkers may be detectable not only in the tumor or its microenvironment but also in the circulation, either as circulating tumor cells (CTCs) or as circulating nucleotides, proteins, or metabolites.
Novel high-throughput molecular technologies that comprehensively characterize the cancer genome and proteome are creating many new possibilities for the identification of biomarkers and, in particular, the development of multimarker (e.g., multigene) panels that integrate information across many markers. Many of the earlier high-throughput approaches such as gene methylation analysis, gene microarrays, comparative genomic hybridization, mass spectrometry/spectroscopy, and bead-based analysis methods are being replaced by next generation sequencing. Gene expression profiles have been explored extensively to identify good and poor prognosis subsets of various human tumors.97 Currently, the ability of methylation analysis to detect early cancer cell DNA methylation aberrations is being investigated. The high-throughput reverse phase protein lysate array (RPPA) proteomic technology allows concurrent analysis of the expression and activation of multiple specific kinases and other proteins.98–102 This platform is particularly suited to investigate kinase signaling in cancer and the molecular effects of novel agents (e.g., TKIs) during treatment and in high-risk tissue during chemoprevention (Figure 1). Emerging mass spectrometry approaches such as MRM (multiple reaction monitoring) and SWATH could provide additional information on candidate genes when high-quality antibodies are not available, and mass spectrometry is particularly useful for biomarker discovery. Together, genomic and proteomic platforms provide information that can be incorporated to develop meta-signatures reflecting global DNA, RNA, and protein abnormalities. These may be capable of outperforming data derived from a single technology examining only one of these platforms.103 For example, proteomic studies can augment genomic panels by providing information on posttranslational modifications and on proteins’ relative levels and activation. As such, data sets, systems for effective data management, integrated analysis of pathways, and “meta-analysis” are critical components of successful development of molecular markers.104–106
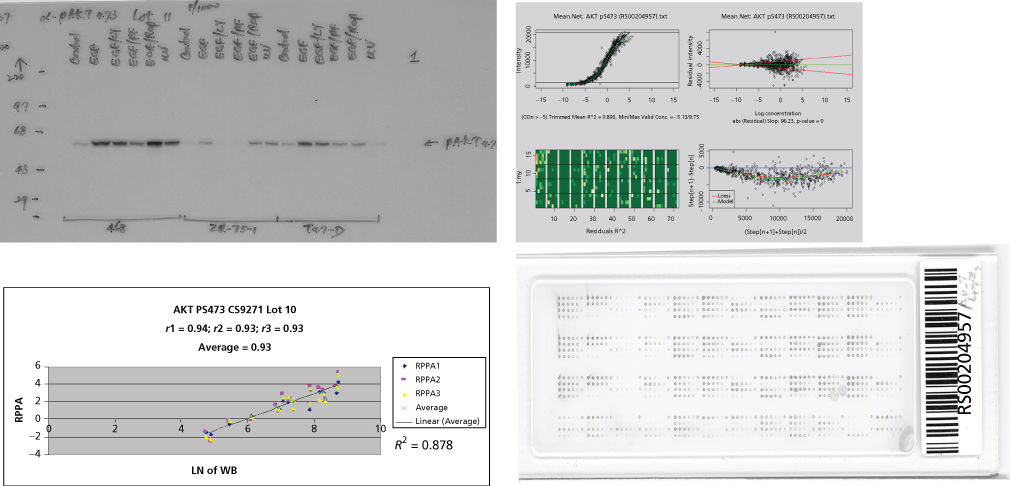
Figure 1 Validation of antibodies for reverse phase protein lysate arrays (RPPAs). Protein lysates from cell lines have been serially diluted on a nitrocellulose-coated slide followed by probing with monospecific antibodies to phosphorylated Akt and signal detection and amplification. Serial dilution curves are used for quantification purposes. Comparisons to western blotting are demonstrated with an r value of 0.878. Quantification and quality control of staining are represented in the upper right panel.
Novel germ line biomarkers
The germ line genome likely contains an underexplored trove of novel cancer risk biomarkers as well as biomarkers for toxicity and efficacy of specific anticancer treatments. For example, germ line polymorphisms have been associated with toxicity and efficacy of the EGFR inhibitor erlotinib in lung cancer.107 Recent large-scale studies of the germ line genome have also begun to uncover a large set of cancer susceptibility markers, many with low penetrance, thereby improving our ability to predict high cancer risk as well as response to therapy utilizing novel germ line biomarkers.108–110
Tissue-specific biomarkers of high risk and for early detection of cancer
Early malignant change may be an indicator of high cancer risk in specific tissues. With availability of less invasive ways to obtain cells likely to be at risk of or to harbor early neoplastic change in tissue at risk of cancer (e.g., in sputum, bronchial washings, blood, feces, urine, or nipple aspirate), the study of tissue markers for screening of carcinogenesis risk and of early malignant transformation becomes more feasible. To date, the cost, invasiveness, lack of large prospective outcome validation studies, and absence of standardized guidelines has confined most of these potentially useful approaches to small clinical studies.
Although screening to detect lung cancer at an early stage using routine cytological examination of sputum did not decrease cancer-specific mortality, the application of molecular detection methods to sputum and bronchial washings is now being studied in an attempt to detect molecular changes associated with premalignant and early malignant bronchial epithelial cells.111 For example, FISH with locus-specific probes to chromosomal regions 5p15, 7p12 (EGFR), 8q24 (C-Myc), and the centromere of chromosome 6 may improve significantly the sensitivity for detection of malignancy in sputum and bronchial washing specimens.112
Cancer-specific biomarkers
As discussed earlier, progress has been made in identifying single biomarkers and multimarker panels (e.g., Oncotype Dx) to predict responses and overcome resistance to targeted therapy. However, progress in this regard has been more limited in other cancer types. Emerging basket clinical trials may address this deficiency in part. Basket trials are based on the observation that common molecular aberrations occur across different cancer types. These trials evaluate the role of targeted therapies against specific molecular aberration(s), regardless of cancer type, within the same trial.113
In the future, pharmacodynamic biomarkers of early drug activity must also be defined in preclinical tumor models and then confirmed in patient samples to assure that patients are receiving a biologically relevant dose of the drug. In this regard, optimal target inhibition by the drug in the tumor may be a more important endpoint than maximum tolerated dose of the drug. However, whether optimal drug activity is dependent on maximal target inhibition, area under the curve, or trough values is not known for most drugs. Precepts derived from Systems Biology such as sensitivity analysis may provide guidelines regarding the optimal inhibition pattern required. This approach may optimize drug efficacy, decrease toxicity, in particular off-target toxicity, and facilitate early identification of nonresponders for triage to alternative therapies. For example, perifosine-induced inhibition of AKT in the tumor correlates remarkably well with tumor growth inhibition using multiple dosing schedules of perifosine. Furthermore, the integrated assessment of the activation status of multiple PI3K/AKT pathway members in the tumor soon after initiation of therapies, using proteomics assays such as RPPA, may prove superior to single markers for prediction of tumor response to PI3K pathway inhibitors.
Serum and urine biomarkers
Novel serum biomarkers have potential utility in cancer screening, in prediction of tumor responsiveness to specific therapies, and in monitoring tumor responses to therapy. As discussed earlier, while conventional serum cancer biomarkers are used routinely for cancer monitoring, their application to screening is limited by suboptimal sensitivity and specificity.114–116 Specificity can be improved by monitoring increases in individual marker levels over time, but marker panels will almost certainly be required to increase sensitivity for screening. The conventional concept concerning the screening utility of serum biomarkers is that their detection should trigger clinical assessment by imaging and biopsy or increased surveillance if appropriate. Alternatively, novel serum markers might be used following screening by other means to increase the specificity of the latter approach. Thus, a novel biomarker may allow definition of an equivocal mammographic lesion as appropriate for serial monitoring or immediate biopsy.
Mass spectrometry-based unbiased approaches to identify novel serum biomarkers from the proteome or metabolome present in blood or urine have the potential to identify biomarker panels that could identify tumors from an early stage of development. Ovarian cancer has been the subject of several such studies because of diagnosis in advanced stages, poor patient outcomes, and the absence of a well-established screening method. Two general approaches have been utilized: identification of distinctive signatures and discovery of discrete markers that might be assembled into panels. However, none of the mass spectrometry-based approaches have validated in large prospectively based sample sets.117
In the urine, the sensitivity of FISH or of cytokeratin (e.g., keratin 19, 20) detection using IHC or reverse transcriptase (RT)-polymerase chain reaction (PCR) may be higher than that of conventional cytology for bladder and urothelial cancer screening.118 A commercial kit (UroVysion) containing hybridization probes for chromosomes 3, 7, 9p21, and 17 is used for FISH analysis of urine. The sensitivity and specificity associated with this analysis were 60% and 82.6%, respectively, for detection of bladder cancer. In contrast, the sensitivity and specificity associated with urine cytology were 24.1% and 90.5%, respectively. Thus, a FISH assay for chromosomes 3, 7, 9, and 17 may have a higher sensitivity than cytology and a similar specificity for the detection of urothelial cancers. Table 2 summarizes the various approaches that have been investigated as potential tools to facilitate bladder cancer screening.119
Table 2 Sensitivity, specificity, and positive predictive value of urine cytology, bladder tumor antigen (BTA) immunoassay, nuclear matrix protein-22 (NMP22) detection, ImmunoCyt, and urine FISH for early detection of bladder cancer
| Sensitivity (%) | Specificity (%) | PPV (%) | |
| PSA | 72 | 93 | 25 |
| Urine cytology | 48–73 | 48–100 | 48–69 |
| BTA | 53 | 77 | 63 |
| NMP22 | 71 | 66 | 21 |
| ImmunoCyt | 78–81 | 74–100 | 26 |
| FISH | 69–71 | 78–95 | 68 |
Note: Serum prostate-specific antigen (PSA) in prostate cancer screening is shown only as a point of reference. ImmunoCyt is currently approved by the US Food and Drug Administration for the monitoring of recurrent bladder cancer. ImmunoCyt uses a cocktail of three monoclonal antibodies to detect bladder cancer cells in the urine. One antibody is directed against a high-molecular-weight form of glycosylated carcinoembryonic antigen, 19A211. The other two antibodies, LDQ10 and M344, are directed against mucins that are specific for bladder cancer and are labeled with fluorescein.
Source: Hu 2007. Reproduced with permission of Oxford University Press.
Circulating tumor cells
CTCs have potential utility in cancer screening, target identification, response prediction, and in monitoring response to treatment. Indeed, the predictive and prognostic utility of CTCs and, more recently, circulating tumor DNA have already been demonstrated in metastatic breast cancer.120, 121 Initial studies of the utility of CTCs in breast cancer screening are proceeding.122 In ovarian cancer, peripheral blood CTC-specific p53 sequences are detectable in some FIGO stage III/IV ovarian cancer patients, suggesting that this approach may be useful as a building block toward early detection.123
The use of CTCs to study molecular biomarkers is limited currently to gene expression signatures because of the need for substrate amplification. However, CTCs have the potential to replace invasive tumor biopsies with “liquid” biopsies and facilitate early access to the tumor genome and proteome for molecular biomarkers predictive of therapy response and resistance, as demonstrated in lung and prostate cancers.124, 125 A major challenge is the difficulty in harvesting CTCs and exploring molecular markers in a limited number of cells. Methods of CTCs enrichment are being explored and include the enhanced density gradient system.126 Currently, we are investigating novel methods of DNA and protein extraction to allow detection of mutations and protein expression/activation changes in CTCs, the latter utilizing RPPA.127, 128
Circulating nucleotides
DNA, RNA, microRNA, and proteins are released from tumor cells and can be found in the circulation. Circulating biomarkers have the potential to reflect processes occurring across all tumor sites in the body that cannot be detected by analysis of the primary tumor and/or metastatic sites. Thus, there is great excitement in the potential for “liquid” biopsies to provide information not accessible from tumor biopsies. Moreover, they would obviate the need for tumor biopsy and, in particular, repeat tumor biopsies to determine tumor response and molecular evolution following therapy. Tumors can release large amounts of nucleotides into the circulation with up to 20% of circulating DNA being derived from a patient tumor. Thus, any aberration such as mutation, rearrangement, increased copy number, increased microRNA, or RNA level could be detected in the circulation, as is the case with KRAS and p53 mutations. However, the amount of DNA in the circulation and the ability to detect tumor-related aberrations varies markedly across tumor types. Nevertheless, commercial tests based on circulating DNA are becoming available.
Challenges in validation of novel molecular biomarkers
The novel unbiased technologies being used to profile the cancer genome or proteome to define new biomarker panels are susceptible to challenges in reproducibility that can be attributed to the simultaneous assay of many gene or protein markers with a limited number of cancer specimens. The large number of potential biomarker combinations introduces a significant likelihood that uncovered associations are simply the result of chance. A rigorous train, test, and validation approach to novel biomarker studies is critical to impact patient management meaningfully. Most studies presenting novel biomarker panels have not impacted patient management to date, either because they did not contend with this multiple parameter problem or because they did not adopt sufficiently robust statistical approaches to validation. The importance of bioinformatic and biostatistical support for the development of novel molecular diagnostics cannot be overemphasized.
Rigorous validation of molecular marker panels in large numbers of well-documented cases is required to test their clinical utility before adoption for routine use in patient management. The applicability of several preliminary panels is still being validated.129, 130 A major hindrance to designing novel molecular studies for biomarker discovery and validation is the frequent lack of availability of adequately preserved and annotated tissue or blood samples in large numbers to correlate with outcome using emerging technologies. In particular, novel high-throughput approaches are often limited in their utility to fresh frozen specimens (paraffin-embedded samples are more plentiful). Thus, a popular model for biomarker development involves discovery using novel high-throughput profiling technologies (e.g., transcriptional profiling) in frozen tissue, followed by validation using moderate-throughput technologies (e.g., RT-PCR) applied to paraffin-embedded tissue. The specific tissue type in which a molecular marker will be validated for clinical use is critical to define.131, 132 Novel comprehensive approaches to biomarker discovery using proteome- or genome-wide expression profiling have now redefined ways tumor banks collect and store tumor samples, with a major emphasis placed on fresh frozen specimens.133, 134
Presently, it is not possible to apply all available investigative technologies to every person, or even every patient at high risk for the development of cancer. Putting cost issues aside, one challenge is to obtain adequate material from biopsy, cellular or serum specimens. Indeed, several hundred nanograms are required for comprehensive analysis of genomic DNA or RNA, but the amount of DNA and RNA available from a fine needle aspirate is in picograms. Furthermore, biopsies frequently have relatively low tumor content, thereby complicating analysis. In the case of CTCs and circulating nucleotides, the DNA and RNA yield can be even smaller. Amplification by PCR can increase mRNA yield, but PCR-generated errors are not uncommon. Approaches to contend with low amounts of protein are minimal. As noted earlier, MRM and other mass spectrometry approaches may complement antibody-based approaches. Currently, we are exploring “barcoding” antibodies with DNA sequences to allow use of amplification approaches that have been applied to DNA detection. These major difficulties need to be addressed to facilitate the routine application of novel molecular technologies to cancer diagnostics.
Recommendations
The effective discovery of novel cancer biomarkers requires the integration of multiple critical factors, including collaborative studies, availability of appropriate human tissue sample sets, standardized reagents and technologies for analyzing, identifying and quantitating candidate biomarkers in tissue and fluid, mouse models of disease, integrated bioinformatics platforms, and implementation of automation, all of which were key to achieving the Human Genome Project, for example. Standard operating procedures (SOPs) regarding tumor specimen biobanking, specimen processing and quality control, validation, performance, and interpretation of analytical assays should be developed at a national or, ideally, international level, to allay concerns regarding potentially unreliable study results and permit accurate pooling and comparison of cross-study results. The American Association for Cancer Research (AACR), the Food and Drug Administration (FDA), the National Cancer Institute (NCI) Cancer Biomarker Collaborative, and the European Group on Tumour Markers have defined these key procedures for effective biomarker development. These strategies can be facilitated by the creation of national biospecimen repositories such as the Cancer Human Biobank, which retrieves highly clinically annotated tumor specimens across centers in the United States and processes them according to SOPs before being made available for analysis.
As biomarker discovery programs characterize tissues using high-throughput genomic, transcriptional and proteomic approaches, the development of adequate and centralized computational infrastructure is critical to allow storage, utilization, and integration of the vast and heterogeneous data derived from novel “omics” technologies. Such a computational resource should be easily accessible to all investigators, protect confidentiality, and avoid duplication of efforts. These resources should facilitate data mining, retrieval, and automated analysis, thus facilitating data integration across molecular platforms and between data sets, and the association of specific aberrations with clinical endpoints. Currency should be maintained as novel “omics” technologies are introduced and upgraded. Access should foster novel biostatistical approaches that further our ability to select clinically useful cancer biomarkers. Approaches are being implemented to facilitate sample and data sharing across the community, given that the amount of data available far exceeds the ability of any group to adequately mine and interpret it. The Biospecimen and Biorepositories Research Branch of the NCI provides effective tools and resources to establish high-quality biobanking. A significant number of online data repositories, such as The Cancer Genome Atlas Portal and The Clinical Proteomic Tumor Analysis Consortium, are now publicly available, thereby facilitating the interrogation of biomarkers by different research groups.
The selection of novel biomarkers from data derived using novel high-throughput technologies needs careful consideration, to discern chance associations from true biological relationships. False biomarker discovery can result from selection bias, overfitting, intrapatient correlation, multiplicity, multiple clinical endpoints, and intrapatient correlation, which must be eliminated from research designs using accurate statistical methods and study designs including sample sizes that enable markers meeting prespecified performance characteristics for well-defined clinical applications to be identified. New biomarkers should be validated before potential introduction into clinical management by measuring the biomarker’s impact on costs and carefully chosen clinical outcomes in a prospective, randomized study.135 However, study costs and regulatory and healthcare market constraints often make such clinical trials impractical. Improved study designs involving restrospective samples, such as the ProBE method, have been developed as alternatives. Alternatively, retrospective analysis of archived prospectively collected samples, or pooled analyses of published and unpublished cohorts with a systematic review can also provide a high level of evidence, as long as specific requirements ensuring high quality within these study designs are met. Moreover, researchers and policymakers turn to simulation modeling to predict the effects of new biomarkers on outcomes. The latter approach can optimize sensitivity, specificity, and cost in addition to identifying leverage points where more definitive biomarkers may be needed. This approach has already been used to assess the cost effectiveness of flexible sigmoidoscopy for colorectal cancer screening.
A Committee on Developing Biomarker-Based Tools for Cancer Screening, Diagnosis, and Treatment of the Institute of Medicine of the National Academies has produced a formal set of recommendations for development of biomarker-based tools for cancer (Box 3).
Box 3 Summary of recommendations to develop biomarker-based tools for cancer
Methods, tools, and resources needed to discover and develop biomarkers
- 1. Federal agencies should develop an organized, comprehensive approach to biomarker discovery and foster development of novel technologies.
- 2. Industry and other funders should establish international consortia to generate and share precompetitive biomarker data.
- 3. Funders should place a major emphasis on developing pathway biomarkers to broaden applicability.
- 4. Funders should sponsor demonstration projects to develop biomarkers that can predict efficacy and safety in patients for drugs already on the market.
- 5. Government agencies and other funders should sustain support for high-quality biorepositories of prospectively collected samples.
- 6. Biomarkers should be developed and validated with high negative predictive value for prediction of response to targeted therapies, particularly those with high cost.
Guidelines, standards, oversight, and incentives needed for biomarker development
- 7. Government agencies and other stakeholders should develop a transparent process to create well-defined consensus standards and guidelines for biomarker development, qualification, validation, and use.
- 8. The FDA and industry should work together to facilitate the codevelopment and approval of diagnostic-therapeutic combinations.
- 9. The FDA should clearly delineate and standardize its oversight of biomarker tests used in clinical decision making.
- 10. The Centers for Medicare and Medicaid Services should develop a specialty area for molecular diagnostics under the CLIA.
Methods and processes needed for clinical evaluation and sdoption
- 11. The Centers for Medicare & Medicaid Services should revise and modernize its coding and pricing system for diagnostic tests.
- 12. The Centers for Medicare and Medicaid Services, as well as other payors, should develop criteria for conditional coverage of new biomarker tests.
As a component of conditional coverage, procedures for high-quality population-based assessments of efficacy and cost effectiveness of biomarker tests should be established.
Source: From the Institute of Medicine of the National Academies.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


