Mutational analysis is important in confirming the diagnosis in patients with clinically evident MEN1 and in identifying first-degree relatives who have inherited, or have not inherited, the MEN1 mutation so that respectively they can either be followed for the development of endocrine tumors or cease to be followed. Half of newly diagnosed index cases are found to have novel mutations, and more than 10% of MEN1 mutations arise de novo and will be passed to subsequent generations.17,19,20 It is important to note that the absence of an MEN1 mutation in a patient with tumors characteristic of MEN1 does not exclude that the patient has MEN1.
The endocrine tumors associated with MEN1 occur more commonly in a sporadic setting, and it is of interest that 25% of gastrinomas, 10% to 20% of insulinomas, 50% of VIPomas, 25% to 35% of bronchial carcinoids, and 20% of parathyroid adenomas express somatic MEN1 mutations. Conversely, somatic MEN1 mutations rarely if ever occur in sporadic adrenocortical tumors, pituitary tumors, or thymus tumors.21
Hereditary Endocrinopathies, Other than MEN1, Involving the Parathyroid and Pituitary Glands
The Parathyroid Gland
Primary hyperparathyroidism occurs in 1 of every 1,000 persons, and 5% to 10% of cases occur in a familial pattern, most often as MEN1, MEN2A, familial hypocalciuric hypercalcemia (FHH), the hyperparathyroidism jaw-tumor syndrome (HPT-JT), and familial isolated hyperparathyroidism (FIHP).
Familial Hypocalciuric Hypercalcemia, Neonatal Severe Hyperparathyroidism, Autosomal Dominant Hypoparathyroidism, and Familial Isolated Hyperparathyroidism
Familial Hypocalciuric Hypercalcemia. The calcium-sensing receptor CASR gene is a seven-transmembrane-spanning G protein-coupled receptor that is expressed in cells of the parathyroid gland and the kidney tubule. CASR plays a critical role in regulating extracellular calcium homeostasis. The discovery of CASR was a surprise, since previously no small cation had been shown capable of acting as a ligand for a G-protein coupled receptor.22 The human CASR, located on chromosome 3q113.3-q21, is sensitive to changes in the ambient calcium concentration and when activated inhibits parathyroid hormone (PTH) secretion and the renal reabsorption of calcium. With heterogeneous inactivating mutations of the CASR gene, the parathyroid cell fails to sense properly an increased serum calcium concentration; the resulting increase in PTH secretion causes FHH (OMIM #145980), an autosomal dominant disease characterized by hypocalciuria, hypercalcemia, mild hypermagnesemia, and parathyroid hyperfunction.23 Bone density levels are not decreased, and patients are generally asymptomatic. It is important for clinicians to recognize this relatively mild form of familial hyperparathyroidism, as it is not cured by parathyroidectomy.24 More than 250 different mutations have been reported to affect CASR function, and mutational analysis is utilized to identify family members who have inherited a mutated CASR allele. It is important to note, however, that CASR mutations are detected in only 65% of patients with FHH and additional mapping studies have revealed additional loci on chromosome 19p and chromosome 19q13.3, thereby suggesting genetic heterogeneity for FHH and raising the possibility that additional or signal pathways are involved in calcium homeostasis.25–27
Neonatal Severe Hyperparathyroidism. With inactivating mutations in both alleles of the CASR gene, neonatal severe hyperparathyroidism (OMIM #239200) develops with serum calcium levels in the range of 15 to 20 mg/dl, up to 10-fold increases in serum PTH levels, greatly enlarged parathyroid glands, demineralization, and bone fractures. The disease was originally described in consanguineous parents, each with FHH. This disease represents a life-threatening emergency, and urgent parathyroidectomy is usually indicated. Recently, however, there are reports that the calcimimetic cinacalcet produces robust and durable reductions in the serum calcium concentrations of patients with neonatal severe hyperparathyroidism. However, the response is not uniform and appears to depend on the patient’s specific CASR mutation.24,28
Autosomal Dominant Hypoparathyroidism. The related syndrome, autosomal dominant hypoparathyroidism (OMIM #146200) results from activating mutations of the CASR gene, causing the parathyroid cells to sense that serum calcium is “elevated” when it is actually normal.29 There is a resulting suppression of PTH secretion (even though it remains within the reference range), enhanced urinary Ca++ secretion, hypomagnesemia, and increased fractional excretion of magnesium. More than 70 activating mutations have been reported in autosomal dominant hypoparathyroidism, and both familial cases and sporadic cases with de novo mutations have been described.30
Familial Isolated Hyperparathyroidism. FIHP (OMIM #146200) is a heterogeneous condition, and some kindreds thought to have this disease have been shown to have germline mutations of MEN1, CASR, CDC73, and CDKN1B suggesting that the disease represents incompletely expressed forms of MEN1, FHH, or the HPT-JT.31 Over 100 families with FIHP have been reported, and in most cases the causative genetic mutation is unknown although there are convincing data that it resides on chromosome 2p13.3-14.32
Hyperparathyroidism-Jaw Tumor Syndrome. HPT-JT (OMIM #145001) is characterized by the autosomal dominant occurrence of hyperparathyroidism, ossifying fibromas of the mandible or maxilla, renal cysts or solid tumors, and uterine fibromas.33 Approximately 50 families with HPT-JT have been reported, and 80 percent of patients have hyperparathyroidism. In 15% of patients with HPT-JT, the parathyroid tumors are malignant. It is noteworthy that the age-related penetrance of HPT-JT is approximately 40% by age 40 years in contrast to MEN1 where the age-related penetrance is 98% by age 40.34 Members of kindreds with this disease need lifelong surveillance by physical examination and biochemical evaluation. It has even been suggested that serial ultrasound examination of the neck should be performed beginning at a young age as parathyroid carcinoma has been reported in normocalcemic family members.35
Inactivating mutations of the HRPT2 tumor suppressor gene, also known as CDC73, causes HPT-JT. The gene is located on chromosome 1q25-q31 and codes for the 531 amino acid tumor suppressor protein, parafibromin (named for parathyroid tumors and jaw fibromas).36 The function of parafibromin is unknown but it is thought to regulate posttranscriptional events and histone modification. There is recent evidence that parafibromin has proaptototic activity, important as a tumor suppressor function.37
The CDC73 gene consists of 17 exons, and the mutations are scattered throughout the coding region, with the majority resulting in functional loss through premature truncation.38 Approximately 100 germline and somatic mutations of CDC73 have been identified but there are no “hot spot” mutations and no relationship between genotype and phenotype.39 Germline CDC73 mutations occur in the majority of patients with the HPT-JT, and surprisingly they also occur in 20% of patients with apparently sporadic parathyroid carcinomas. Somatic CDC73 mutations have been detected in 50% to 100% of patients with sporadic parathyroid carcinoma.40,41 Mutations in CDC73 are rarely seen in sporadic parathyroid adenomas, an important diagnostic finding distinguishing benign from malignant parathyroid tumors.37,42
The Pituitary Gland
Familial Isolated Pituitary Adenomas. Approximately 5% of pituitary tumors occur in a familial pattern. Familial isolated pituitary adenomas (FIPA [OMIM #102200]) is an autosomal dominant disease characterized by the occurrence of at least two cases of pituitary tumors in a family that has no features of MEN1 or the Carney complex.43 In a study of 64 families with FIPA, 55 of 138 affected family members had prolactinomas, 47 had somatotropinomas, 28 had nonsecreting adenomas, and eight had adrenocorticotropic hormone–secreting tumors.44 There are two equally occurring phenotypes, where families are either homogenous, expressing the same type of pituitary tumors, or heterogeneous, expressing different types of pituitary tumors. In heterogeneous families, prolactinomas exhibit more aggressive behavior, compared to their sporadic counterparts, with higher rates of subsella expansion and cavernous sinus invasion. Conversely, somatotropinomas are more aggressive in the homogenous setting.
Germline mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene occur in 20% (range 15% to 40%) of patients with FIPA; however, in the remainder, the causative gene is unknown.45 The AIP gene consists of six exons encoding a 330 amino acid (37 kDa) cytoplasmic co-chaperone phosphoprotein (XAP2).46 The gene is located at 11q13, which is 2.7 Mb downstream from the MEN1 gene. AIP forms a complex with the aryl hydrocarbon receptor and two 90-kD heat-shock proteins (HSP90). The aryl hydrocarbon receptor is a ligand-activated transcription factor and also participates in cellular signaling pathways.47 The mean age at diagnosis is 13 years earlier in AIP-positive FIPA families compared to AIP-negative families, and the majority of patients in AIP-positive families are male, where there is equal gender balance in AIP-negative families. The pituitary tumors in AIP-positive patients are large, frequently invade surrounding structures, and respond poorly to somatostatin analogues.48,49
Multiple Endocrine Neoplasia Type 2
Multiple Endocrine Neoplasia Type 2A and Multiple Endocrine Neoplasia 2B
Clinical Features. In 1968, Steiner and associates described a large family with medullary thyroid carcinoma (MTC), pheochromocytomas (PHEO), hyperparathyroidism (HPTH), and Cushing syndrome.50 The disease was named MEN type 2, and we now it as MEN2A. Until recently, there were three related syndromes of hereditary MTC: MEN2A (OMIM #171400), MEN2B (OMIM #162300), and familial MTC (FMTC) (OMIM #155240).51,52 Most endocrinologists now feel that FMTC represents a variant of MEN2A and should not stand alone as a disease entity. It has recently been suggested that there be two MEN2 syndromes: MEN2A and MEN2B. In addition, within MEN2A, it has been suggested that there be four variants: classical MEN2A, MEN2A with Hirschsprung disease, MEN2A with cutaneous lichen amyloidosis, and FMTC.53
Of patients with hereditary MTC, 95% have MEN2A and 5% have MEN2B. Virtually all patients with these syndromes develop MTC, and up to half of the patients with MEN2A and MEN2B develop PHEOs. The PHEOs are almost always benign and confined to the adrenal gland. Patients with a unilateral PHEO usually develop a contralateral PHEO within 10 years.54 Prior to the development of biochemical and genetic test to establish the early diagnosis of hereditary MTC, PHEO, not MTC, was the most common cause of death in patients with MEN2A.55 Approximately 30% of patients with MEN2A develop HPTH, which is usually mild and asymptomatic.
Patients with MEN2B have a typical physical appearance with a Marfanoid habitus and atypical facies. They also have a generalized ganglioneuromatosis and ocular and skeletal abnormalities.
MTC originates from the neural crest–derived C-cells. The MTC cells secrete the polypeptide hormone calcitonin (CTN) and the glycoprotein carcinoembryonic antigen. Serum levels of CTN and carcinoembryonic antigen are excellent tumor markers, most useful in detecting persistent or recurrent disease following thyroidectomy for MTC. The most effective therapy for MTC is thyroidectomy, preferentially performed before MTC develops or while it is still confined to the thyroid gland.
Molecular Genetics of MEN2A and MEN2B. In 1985, Takahashi and associates discovered the RET (REarranged during Transfection) protooncogene.56 The gene is located in the pericentromeric region of chromosome 10q11.2 and includes 21 exons. RET encodes a receptor tyrosine kinase, which is expressed in neuroendocrine cells (including thyroid C cells and adrenal medullary cells), neural cells (including parasympathetic and sympathetic ganglion cells), urogenital tract cells, and branchial arch cells. RET is essential for the development, survival, and regeneration of neuronal cells in the gut, the kidney, and the nervous system.
The RET gene has an extracellular portion containing four cadherin-like repeats, a calcium binding site and a cysteine–rich region, a transmembrane portion, and an intracellular portion containing two tyrosine kinase domains.53 Alternate splicing of RET produces three isoforms with either 9, 43, or 51 amino acids at the C terminus, referred to as RET9, RET43, and RET51.57,58 Mice lacking RET51 are normal; however, mice lacking RET9 have renal malformation and defects in innervation of the gut.59 RET51 and RET9 have transforming activity but RET51 is the stronger of the two.
A tripartite complex is necessary for RET signaling. One of four glial-derived neurotrophic factors (GDNF) family ligands; GDNF, neurturin, persephin, or artemin binds RET in conjunction with one of four glycosylphosphatidylinositol-anchored co-receptors, designated GDNF family receptors (GFR): GFR-α1, GFR-α2, GFR-α3, or GFR-α4.60–62 The GDNF family ligand–GFR complex causes dimerization of RET with activation of autophosphorylation and intracellular signaling. The molecular pathways associated with activation of the RET protooncogene are shown in Fig. 81.2.53
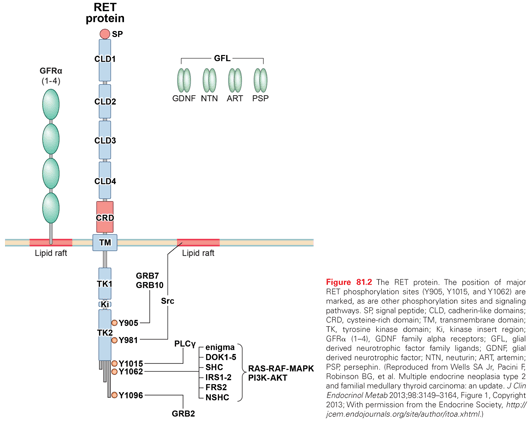
MEN2A and MEN2B are caused by mutations in the RET protooncogene; the most common are shown in Fig. 81.3.63,64 In approximately 50% of patients with MEN2B and 10% of patients with MEN2A, the RET mutation arises de novo and virtually always from the paternal allele. Furthermore, in MEN2B there is a gender preference such that the first offspring of transmitting males is almost always an affected female.65
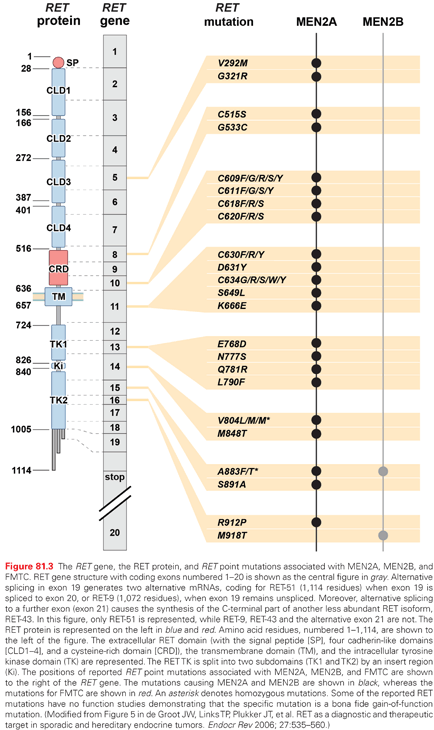
In patients with MEN2A, there is a correlation between genotype and phenotype. In 98% of patients with MEN2A, mutations occur in RET codons 609, 611, 618, 620, and 634. RET codon 634 accounts for 80% of mutations in MEN2A. The frequency of PHEO depends on the RET codon mutation: (634 [50%], 609 [4% to 26%], 611 [10% to 25%], 618 [12% to 23%], and 620 [13% to 24%]).66,67 A RET codon 634 mutation is associated with a 30% incidence of HPTH; however, the frequency ranges between 2% and 10% in patients with mutations in RET codons 609, 611, 618, and 620.68 Patients with MEN2A may also develop cutaneous lichen amyloidosis, which is almost always associated with a RET codon 634 mutation.69 Less commonly, patients with MEN2A develop Hirschsprung disease, which is associated with RET codon mutations in exon 10: 609 (15%), 611 (5%), 618 (30%), and 620 (50%).70
Approximately 95% of patients with MEN2B have mutations in RET codon M918T, and most of the remainder have mutations in RET codon A833F.71,72 Rarely, patients with MEN2B have double mutations appearing in tandem on the same allele involving RET codon V804M and either codon Y806C, S904C, E805K, or Q781R. The MEN2B syndrome in patients with these double mutations is atypical, and MTC presents at a later age than in patients with typical MEN2B.73–76 Approximately half of the patients with sporadic MTC have a somatic RET M918T mutation, which it appears is associated with a more aggressive clinical phenotype.77 Recently, it was discovered that the majority of patients with sporadic MTC who have no somatic RET mutations have somatic mutations of HRAS, KRAS, or rarely NRAS.78 There is no indication that the presence of somatic RAS mutations is associated with a pattern of clinical behavior of the MTC.
Of patients with presumed sporadic MTC, 7% will be found on direct DNA analysis to have germline RET
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


