INTRODUCTION
SUMMARY
The coagulation cascade consists of a complex network of reactions that are essential for the conversion of zymogens into enzymes and of inactive procofactors into cofactors. Most of these reactions take place on a membrane surface, which restricts coagulation to the site of injury. Upon initiation, these reactions serve to produce the fibrin that is necessary for the formation of a stable hemostatic plug. In addition, these reactions provide feedback loops that limit and localize thrombus formation and regulate thrombus resolution. This chapter highlights key biochemical characteristics of the individual coagulation factors, essential aspects regarding their synthesis, and the clinical importance of acquired or inherited variations that affect their quantity or function. The coagulation factors are grouped as (1) the vitamin K-dependent zymogens (prothrombin, factor VII, factor IX, factor X, and protein C); (2) the procoagulant cofactors (factor V, factor VIII); (3) the soluble cofactors (protein S, von Willebrand factor); (4) factor XI and the contact system (factor XII, prekallikrein, and high-molecular weight kininogen); (5) the cell-associated cofactors (tissue factor, thrombomodulin, endothelial protein C receptor); (6) the fibrin network (fibrin[ogen], factor XIII, thrombin-activatable fibrinolysis inhibitor); and (7) inhibitors of coagulation (antithrombin, tissue factor pathway inhibitor, protein Z/protein Z-dependent protease inhibitor). Table 113–1 summarizes the major features of the coagulation factors addressed in this chapter. The final sections of this chapter present an overview of the coagulation cascade in which the pathways of hemostasis including the contribution of endothelial cells, blood platelets, and immune cells are described.
Acronyms and Abbreviations
APC, activated protein C; ADP, adenosine diphosphate; AT, antithrombin; C4BP, complement 4b–binding protein; COX, cyclooxygenase; EGF, epidermal growth factor; EPCR, endothelial protein C receptor; ER, endoplasmatic reticulum; ERGIC, ER-Golgi intermediate compartment; Gla, γ-carboxy glutamic acid; GP, glycoprotein; HK, high-molecular-weight kininogen; LMAN1, mannose-binding lectin-1; PAI-1, plasminogen activator inhibitor type 1; PAR, protease-activated receptor; PK, prekallikrein; poly-P, polyphosphate; RVV, Russell’s viper venom; SHBG, sex hormone–binding globulin; TAFI, thrombin-activatable fibrinolysis inhibitor; TFPI, tissue factor pathway inhibitor; UFH, unfractionated heparin; VWD, von Willebrand disease; VWF, von Willebrand factor; ZPI, protein Z–dependent protease inhibitor.
| Protein | Plasma Concentration | Mr | Plasma Half-Life | ||
|---|---|---|---|---|---|
| (μg/mL) | (nmol/L) | (kDa) | (Hours) | ||
| ZYMOGENS | |||||
| + Gla domain | Prothrombin (factor II) | 100 | 1400 | 72 | 60 |
| Factor VII | 0.5 | 10 | 50 | 3–6 | |
| Factor IX | 5 | 90 | 55 | 18–24 | |
| Factor X | 10 | 170 | 59 | 34–40 | |
| Protein C | 4 | 65 | 62 | 6–8 | |
| – Gla domain | Factor XI | 5 | 30 | 160 | 60–80 |
| Factor XII | 40 | 500 | 80 | 50–70 | |
| Prekallikrein | 40 | 490 | 85 | 35 | |
| Factor XIIIA*† | – | – | 83 | – | |
| Factor XIIIB* | 7 | 94 | 76.5 | – | |
| Factor XIII | 30 | 94 | 320 | 240 | |
| TAFI | 4–15 | 70–275 | 60 | – | |
| COFACTORS | |||||
| Soluble | Factor V† | 5-10 | 20 | 330 | 12–36 |
| Factor VIII | 0.2 | 0.7 | 300 | 8–12 | |
| VWF | varies | 10 | 500–20,000 | 8–12 | |
| Protein S‡ | 25 | 350 | 75 | 42 | |
| Protein Z§ | 2.5 | 40 | 62 | 60 | |
| HK | 80 | 670 | 120 | 150 | |
| Cellular | Tissue factor | – | – | 47 | – |
| Thrombomodulin | – | – | 78 | – | |
| EPCR | – | – | 49 | – | |
| STRUCTURAL PROTEIN | Fibrinogen | 2500 | 7400 | 340 | 72–120 |
| Aα chain | – | – | 66.5 | – | |
| Bβ chain | – | – | 52 | – | |
| γ Chain | – | – | 46.5 | – | |
| INHIBITORS | Antithrombin | 150 | 2500 | 58 | 60–72 |
| TFPIᶠ| 0.01 | 0.25 | 40 | 0.03 | |
| ZPI§ | 4 | 60 | 72 | 60 | |
MOLECULAR BIOLOGY AND BIOCHEMISTRY OF THE COAGULATION FACTORS
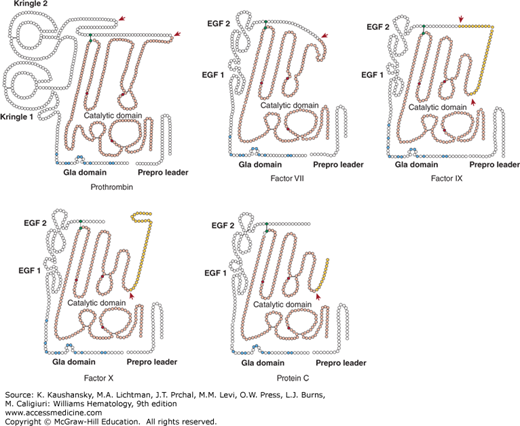
The vitamin K-dependent zymogens circulate in an inactive state and require proteolytic activation to function as a serine protease. All share a similar domain structure of a C-terminal serine protease domain and an N-terminal γ-carboxy glutamic acid (Gla) domain, which are connected by two epidermal growth factor (EGF)-like domains or kringle domains (Fig. 113–1). Each protein domain has a well-defined function and facilitates substrate recognition, interaction with protein cofactors, or binding to a negatively charged lipid surface, such as that of activated platelets or endothelial cells, thereby restricting coagulation to the site of injury. The latter is mediated via the Gla domain, a domain that is characteristic to the vitamin K–dependent proteins.
Figure 113–1.
Vitamin K–dependent schematic of the vitamin K–dependent zymogens. Each circle represents an amino acid. The prepro leader sequence contains the signal peptide as well as the propeptide that directs γ-carboxylation of glutamic acid (Gla) residues. Cleavage of the prepro sequence from the mature protein is indicated by the separation between the two. The Gla domains are indicated with the Gla residues in blue. Prothrombin has a finger loop followed by two kringle domains. Factors VII, IX, X, and protein C have epidermal growth factor (EGF)-like domains. Prothrombin, factor VII, and factor IX circulate as single-chain molecules. Factor X and protein C circulate as two chains that are disulfide linked. All have homologous serine protease (“catalytic”) domains (shown in light red), in which the active site His, Asp, and Ser residues are indicated in dark red. Cleavages that convert the zymogen to an active enzyme are indicated by the red arrows. In factor IX, factor X, and protein C, the released activation peptide is indicated in yellow. After proteolytic activation, all of the molecules are two-chain disulfide-linked molecules, with the cysteines forming a disulfide bridge (black line) indicated in green. All catalytic domains but that of prothrombin remain attached to the Gla domain following activation.
The high level of protein and gene homology suggests that the vitamin K–dependent zymogens originate from a common ancestral gene as a result of gene duplications.1 Exon shuffling and tandem duplication may account for the generation of the ancestral gene, in which the functional domains that are encoded by a single exon each were combined and duplicated.2 This process may also account for the presence of the kringle domains as opposed to EGF-like domains in prothrombin.
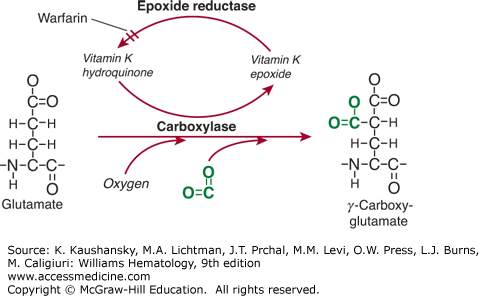
The Gla domain refers to the 42-residue region located in the N-terminus of the mature protein that comprises 9 to 12 glutamic acid residues that are posttranslationally γ-carboxylated into Gla residues by a specific γ-glutamyl carboxylase in the endoplasmatic reticulum of hepatocytes.3 This γ-carboxylase requires oxygen, carbon dioxide, and the reduced form of vitamin K for its action, hence the name vitamin K–dependent proteins. For each Glu residue that is carboxylated, one molecule of reduced vitamin K is converted to the epoxide form (Fig. 113–2). Vitamin K epoxide reductase converts the epoxide form of vitamin K back to the reduced form.4 Warfarin and related 4-hydroxycoumarin–containing molecules inhibit the activity of vitamin K epoxide reductase, thereby preventing vitamin K recycling and inhibiting γ-carboxylation. This results in a heterogeneous population of circulating undercarboxylated forms of the vitamin K–dependent proteins with reduced activity. Because warfarin blocks the reductase and not the carboxylase, the inhibitory effect of warfarin can be (temporarily) reversed by administration of vitamin K. Recognition by and interaction with γ-carboxylase is facilitated by the propeptide sequence that is located C-terminal to the signal peptide. The propeptide is highly conserved among the vitamin K–dependent proteins, and amino acids at positions –18, –17, –16, –15, and –10 are critical to recognition by the γ-carboxylase.5,6 Following γ-carboxylation, the propeptide is removed through limited proteolysis prior to secretion of the mature protein.
Figure 113–2.
Vitamin K–dependent γ-carboxylation. Glutamic acid residues are converted to γ-carboxy glutamic acid (Gla) residues by a specific γ-carboxylase. This reaction requires oxygen, carbon dioxide (shown in green), and reduced vitamin K in the form of a hydroquinone. Carbon dioxide is incorporated onto the γ-carbon, providing a second carboxylate group on that residue. In the process of this reaction, reduced vitamin K is converted to an epoxide. Reduced vitamin K is recycled by a specific epoxide reductase, a reaction that can be blocked by warfarin and warfarin analogues.
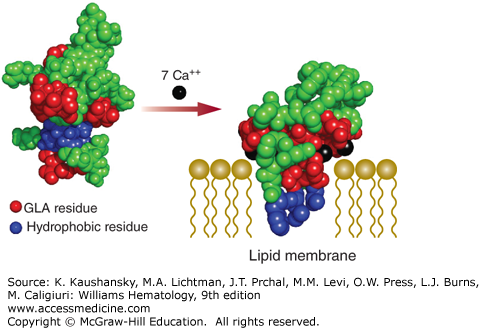
A correctly γ-carboxylated Gla domain is essential for interaction of the vitamin K–dependent proteins with phosphatidylserine, a negatively charged phospholipid. Under normal conditions, phosphatidylserine is not exposed on the outer membrane leaflet of cells. However, in activated endothelial cells or platelets, phosphatidylserine is part of the extracellular cell surface where it supports blood coagulation reactions. The Gla domain interacts with the anionic cell surface in a calcium-dependent manner. These calcium ions are coordinated by Gla residues and induce a conformational change in the Gla domain that is characterized by the appearance of a hydrophobic surface loop (Fig. 113–3). Membrane binding by the Gla domain occurs when this hydrophobic surface loop penetrates into the hydrophobic portion of the phospholipid bilayer, which is facilitated by the interaction of the Gla-bound calcium ions with the phosphate head groups of phosphatidylserine.7,8 It has been shown that the phosphate head groups of exposed phosphatidylethanolamine are also capable of coordinating calcium ions, thereby contributing to the interaction of the Gla domains with the negatively charged membrane surface.9
Figure 113–3.
Calcium-dependent association of the glutamic acid (Gla) domain with the anionic phospholipid surface. Non–calcium bound [Protein Data Bank (PDB) structure 2PF2] and calcium bound (PDB structure 1WHE) molecular models of the Gla domain of prothrombin are shown. Circles represent amino acids, with the Gla (GLA) residues indicated in red. Hydrophobic residues involved in membrane insertion are shown in blue. In the absence of calcium, the negatively charged Gla residues are exposed to the solution and the hydrophobic residues are buried. Association of calcium ions (in black) to the Gla residues provides sufficient energy to alter the overall conformation of the Gla domain and expose the hydrophobic residues. Membrane binding by the Gla domain occurs when this hydrophobic surface loop penetrates into the hydrophobic portion of the phospholipid bilayer (drawn schematically), which is facilitated by interaction of the Gla-bound calcium ions with the negatively charged phosphate head groups.
The serine protease domains of the vitamin K–dependent proteins are highly homologous, as they bear a chymotrypsin-like fold and display trypsin-like activity.10 Once activated, they cleave peptide bonds following a positively charged amino acid (Lys or Arg). Activation proceeds through proteolysis at one or more sites N-terminal to the serine protease domain (see Fig. 113–1). Subsequently, the newly formed N-terminus inserts into the serine protease domain to form a salt bridge with an Asp residue, which is associated with conformational changes in the serine protease domain. These lead to an optimal configuration of the active site through alignment of the active site residues His, Ser, and Asp, and to formation of the substrate-binding exosites, allowing for substrate conversion. The substrate-binding exosites are unique to each vitamin K–dependent protease and are responsible for their highly specific substrate recognition and associated function in coagulation.
Interaction of the vitamin K–dependent proteases with specific cofactors on a (anionic) membrane surface (Table 113–2) further enhances substrate recognition, as the cofactors interact with both the protease and the substrate, bridging the two together. This results in a dramatic enhancement of the catalytic activity (Table 113–3), thereby making the cofactor–protease complex the physiologic relevant enzyme. The increase in catalytic rate has also been attributed to a cofactor-induced conformational change in the protease.11 However, whether this molecular mechanism holds true for all cofactor–protease complexes remains to be determined. Tissue factor is the cofactor for factor VIIa, factor VIIIa is the cofactor for factor IXa, and factor Va is the cofactor for factor Xa, while thrombin does not require a cofactor for its procoagulant activity. However, upon association with the cofactor thrombomodulin, thrombin’s specificity is changed from procoagulant to anticoagulant (cleaving and activating protein C). The complexes are also named for their physiologic substrate: the factor VIIIa–factor IXa complex is termed the “tenase” or “intrinsic tenase” complex; the tissue factor–factor VIIa complex is termed the “extrinsic tenase” complex; and the factor Va–factor Xa complex is termed the “prothrombinase” complex.
| Protease | Cofactor | Substrate | Cellular Location |
|---|---|---|---|
| Factor VIIa | Tissue factor | Factor IX | Many cells* |
| Factor X | |||
| Factor IXa | Factor VIIIa | Factor X | Platelets |
| Factor Xa | Factor Va | Prothrombin | Platelets |
| Thrombin | Thrombomodulin | Protein C | Endothelium |
| Activated protein C | Protein S | Factor Va Factor VIIIa | Endothelium |
Prothrombin, or factor II, which was discovered by Pekelharing in 1894, is one of the four coagulation factors that were described by Paul Morawitz in 1905, in addition to fibrinogen (factor I), thromboplastin (thrombokinase, factor III, now tissue factor), and calcium (factor IV).12,13 The zymogen prothrombin is primarily synthesized in the liver and circulates in plasma as a single-chain protein of 579 amino acids (Mr ≈72,000) at a concentration of 1.4 μM with a plasma half-life of 60 hours (see Table 113–1).
Prothrombin is composed of fragment 1 (F1), fragment 2 (F2), and the serine protease domain. F1 consists of the Gla domain, which comprises 10 Gla residues, and the kringle 1 domain; F2 contains the kringle 2 domain (see Fig. 113–1). The two kringle domains, which replace the EGF-like domains present in most vitamin K–dependent zymogens, are conserved secondary protein structures that fold into large loops that are stabilized by three disulfide bonds and schematically resemble a Danish pastry called a “kringle.” Their primary function is to bind other proteins such as the cofactor Va and serine protease factor Xa that activate prothrombin.
Other than γ-carboxylation of Glu residues, prothrombin is posttranslationally modified via N-glycosylation in the kringle 1 (Asn78, Asn143) and serine protease domains (Asn373), which contributes to the stability of the prothrombin precursor during processing in the endoplasmatic reticulum.14,15
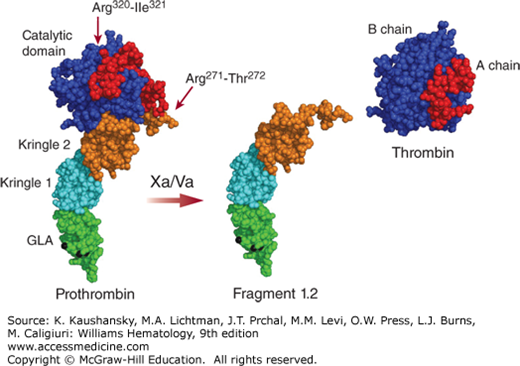
Prothrombin is proteolytically activated by the prothrombinase complex (i.e., factor Va, factor Xa, calcium, and anionic phospholipids) that cleaves at Arg271 and Arg320 (see Fig. 113–1). Cleavage at Arg320 opens the active site of the protease domain, while cleavage at Arg271 removes the activation fragment F1.2 (F1.2). Both cleavages are necessary to generate procoagulant α-thrombin (IIα) (Fig. 113–4). The composition of the membrane surface directs the cleavage order in prothrombin and the formation of either the zymogen prethrombin 2 (initial cleavage at Arg271) or the proteolytically active intermediate meizothrombin (initial cleavage at Arg320).16,17 Meizothrombin has impaired procoagulant activity as compared to α-thrombin, but superior anticoagulant activity as it displays increased thrombomodulin-dependent protein C activation, which is likely facilitated by membrane binding of meizothrombin through its Gla domain.18 The snake venom protease Ecarin is capable of generating meizothrombin specifically through proteolysis at Arg323 only. However, this meizothrombin is instable as a result of autocatalysis at Arg155, thereby removing the Gla domain containing F1. The so formed meizo-des-F1 can be converted to thrombin by prothrombinase, but at a slower rate as it is incapable of membrane binding. Assessment of F1.2 levels reflects prothrombin activation and is commonly used as a marker for thrombin generation.
Figure 113–4.
Prothrombin to thrombin conversion. A molecular model of prothrombin comprising the γ-carboxy glutamic acid (Gla; GLA) domain, both kringle domains, and the catalytic domain is shown (PDB structures 2PF2, 1HAG, 1A0H, 1HAI). Gla domain-bound calcium ions are indicated in black. Cleavage by the factor Va-Xa complex at Arg271 and Arg320 releases thrombin (with the A chain and catalytically active B chain) from the rest of the molecule (fragment 1.2).
Thrombin (IIα) is a two-chain serine protease (Mr ≈37,000) comprising a light chain of 49 residues (A chain; Mr ≈6000) that is covalently linked to the catalytic heavy chain of 259 residues (B chain; Mr ≈31,000). Thrombin’s main function is to induce the formation of a fibrin clot by removing fibrinopeptides A and B from fibrinogen to form fibrin monomers, which then spontaneously polymerize. In addition, thrombin is able to cleave a wide variety of substrates with high specificity, which is mediated via its negatively charged, deep active site cleft and via the anion binding exosites I and II that specifically interact with cofactors and/or substrates.19 The dynamic structural conformation of thrombin allows for binding to diverse ligands, and the subsequent ligand-induced conformational stabilization, known as thrombin allostery, regulates and controls thrombin activity.20,21
Thrombin initiates important procoagulant pathways by proteolytic activation of the cofactors V and VIII and zymogen factor XI that collectively amplify thrombin and fibrin formation, and by activating factor XIII that crosslinks and stabilizes the fibrin polymers. Another procoagulant function of thrombin is to inhibit fibrinolysis by proteolytic activation of the thrombin-activatable fibrinolysis inhibitor (TAFI), a reaction enhanced by the endothelial-bound cofactor thrombomodulin. Thrombin also has an anticoagulant function and upon binding to the cofactor thrombomodulin, it is capable of proteolytically activating protein C, which inactivates the cofactors Va and VIIIa.
Thrombin activates the seven-transmembrane domain, G-protein–coupled protease-activated receptors (PARs) PAR1, PAR3, and PAR4 that are expressed on a wide range of cell types in the vasculature by proteolytic cleavage of their N-terminal extracellular domains.22,23,24,25 Thrombin is one of the strongest platelet activators in vivo and activates platelet-expressed PAR1 and PAR4.25 The platelet glycoprotein (GP) Ib (GPIb) serves as a cofactor for thrombin in PAR1 cleavage (Chap. 112). Thrombin-mediated activation of endothelial-PAR1 triggers release of von Willebrand factor (VWF) and P-selectin, which promote rolling and adhesion of platelets and leukocytes. In addition, this stimulates the endothelial production of platelet-activating factor, a potent platelet and leukocyte activator, as well as the production of chemokines, cyclooxygenase (COX)-2, and prostaglandins.25 Thrombin-mediated PAR activation is not only critical for coagulation, but also plays an important role in inflammatory and proliferative responses associated with vascular injury, such as in atherosclerosis and cancer.26
The physiologic inhibitors of thrombin are the serine protease inhibitors (serpins) antithrombin, heparin cofactor II, protein C inhibitor, and protease nexin 1, with antithrombin being the primary plasma inhibitor. For all four serpins, the rate of thrombin inhibition can be accelerated by glycosaminoglycans, such as heparin (Table 113–4), through mutual binding to the serpin and thrombin (see Fig. 113–2), which ensures rapid inhibition of thrombin at the intact endothelial cell surface where heparin-like glycosaminoglycans are found.
| Second-Order Association Rate Constants (M–1s–1) | |||
|---|---|---|---|
| Protease | – Heparin | + H5 | + UFH |
| Thrombin | 7.7 × 103 | 1.5 × 104 | 4.7 × 107 |
| Factor Xa | 2.6 × 103 | 7.6 × 105 | 6.6 × 106 |
| Factor IXa | 58 | 3.1 × 104 | 6.2 × 106 |
| TF-Factor VIIa | 33 | 4.9 × 103 | 1.5 × 104 |
| Factor Xia | 3.6 × 102 | 1.1 × 103 | 1.8 × 105 |
| Factor XIIa | 39 | 1.9 × 103 | 6.6 × 104 |
| APC | 0.08 | 1.9 | 2.1 |
Heparin and heparin-derivatives are clinically used as anticoagulants to inhibit thrombin via antithrombin. Hirudin, which originates from the salivary glands of medicinal leeches, and its recombinant and synthetic derivatives are potent and highly specific inhibitors that directly target the active site and exosite I of thrombin.27 The target-specific oral anticoagulant dabigatran also inhibits thrombin directly with high specificity and reversibly binds the active site of thrombin.27,28
Prothrombin is encoded by a gene (F2) on chromosome 11p11.2 that is approximately 20 kb long.29 The coding sequence is divided over 14 exons that range in size from 25 to 315 bp (Fig. 113–5). The reference sequence of prothrombin mRNA comprises 2018 bases. There are no common, well characterized, splicing variants with known biology.
Figure 113–5.
Relationship of gene structure to protein structure in prothrombin. The exons, introns, mRNA, and protein structure are as indicated. The mRNA is 2 kb with small 5′ and 3′ untranslated regions (shown in light blue). In the protein, Pro indicates the prepro leader sequence, GLA indicates the γ-carboxy glutamic acid (Gla) domain, Kringles 1 and 2 are indicated, LC indicates the light chain (also known as the A chain), and the serine protease (catalytic) domain is indicated.
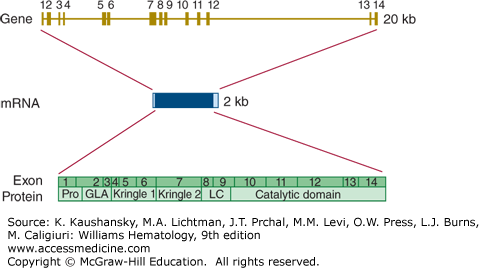
Homozygosity or compound heterozygosity for loss of function mutations in the prothrombin gene leads to a bleeding tendency. This condition is quite rare with perhaps one case per 2,000,000 newborns.30 Heterozygous carriers of loss-of-function mutations are without a bleeding phenotype. Mutations have been characterized in a relatively small number of cases with homozygous or compound heterozygous prothrombin deficiency (consult the human gene mutation database at http://www.hgmd.org for details). The majority of mutations underlying prothrombin deficiency are missense mutations, but several small deletions/insertions have also been reported.
Gain-of-function mutations in the prothrombin gene increase thrombotic risk. The best known variation is G20210A.31 This variation of the last nucleotide preceding the poly(A)-tail of the mature mRNA has an effect on 3′-end mRNA processing and increases the level of prothrombin in plasma by approximately 10 to 20 percent in heterozygous individuals.32 This relatively small increase in the level of prothrombin results in a two- to threefold enhanced risk for venous thrombosis. Homozygotes for the G20210A variation are quite rare, and the risk associated with homozygosity has not been measured with certainty. The G20210A variation is relatively common in whites, with a strong south-north gradient in that the variation is most common in southern Europe.33
Factor VII, which was discovered around 1950,34 is synthesized in the liver and circulates in plasma as a single-chain zymogen of 406 amino acids (Mr ≈50,000) at a concentration of 10 nM with a short plasma half-life of 3 to 6 hours (see Table 113–1).
Factor VII consists of a Gla domain with 10 Gla residues, two EGF-like domains, a connecting region, and the serine protease domain (see Fig. 113–1). Calcium coordination in EGF-1 is mediated by partial hydroxylation of Asn63 and O-linked fucosylation of Ser60.35 Further posttranslational modifications of factor VII consist of O-linked (Ser52 in EGF-1) and N-linked (Asn154 in the connecting region, Asn322 in the serine protease domain) glycosylation.
Factor VII is proteolytically activated once it has formed a high-affinity complex with its cofactor tissue factor. A number of coagulation proteases including thrombin and factors IXa and XIIa are capable of cleaving factor VII at Arg152 to generate factor VIIa (see Fig. 113–1), with factor Xa being considered the most potent and physiologically relevant activator of factor VII.36 Autoactivation can also occur, which is initiated by minute amounts (approximately 0.1 nM) of preexisting factor VIIa.37
Factor VIIa is a two-chain serine protease composed of a light chain (Mr ≈20,000) comprising the Gla and EGF domains and the catalytic heavy chain (Mr ≈30,000), which are covalently linked via a disulfide bond. Factor VIIa activity is only expressed when bound to tissue factor, which induces an active conformation of the factor VIIa serine protease domain (Fig. 113–6).11 Factor VIIa interacts with tissue factor via its Gla and EGF domains.
Figure 113–6.
The tissue factor–factor VIIa complex. A molecular model of factor VIIa (PDB structures 1QHK, 1WHF, 1RFN, 1DAN) and the crystal structures of the tissue factor–factor VIIa complex (PDB structure 1DAN) and the extracellular domain of tissue factor (PDB structure 2HFT) are shown. The γ-carboxy glutamic acid (Gla; GLA) domain, epidermal growth factor (EGF)-like domains 1 and 2, and serine protease (catalytic) domain of factor VIIa are indicated. Binding to tissue factor alters the overall structure of factor VIIa.
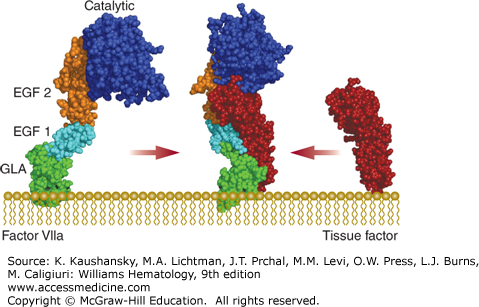
The tissue factor–factor VIIa complex activates both coagulation factors IX and X, which is considered to be the main initiating step of the extrinsic pathway of coagulation. In addition, the tissue factor–factor VIIa (–factor Xa) complex is not only critical to processes in coagulation, but also to wound healing, angiogenesis, tissue remodeling, and inflammation through proteolytic activation of PAR2.38,39,40
The ternary tissue factor–factor VIIa–factor Xa complex is inhibited by the tissue factor pathway inhibitor (TFPI). Tissue factor–factor VIIa is also inhibited by antithrombin, but only in the presence of heparin (see Table 113–4).
The gene encoding factor VII (F7) is located on chromosome 13q34, is almost 15 kb in length, and comprises 9 exons (Fig. 113–7). The canonical mRNA encoding factor VII comprises 3000 bases.41 Alternatively spliced transcript variants encoding multiple isoforms have been observed, but their biology is not well characterized.42
Figure 113–7.
Relationship of gene structure to protein structure in factor VII. The exons, introns, mRNA, and protein structure are as indicated. The mRNA is 2.7 kb with a small 5′ untranslated region and a relatively large 3′ untranslated region (light blue). In the protein, Pro indicates the prepro leader sequence, GLA indicates the γ-carboxy glutamic acid (Gla) domain, and epidermal growth factor (EGF)-1 and -2, as well as the serine protease (catalytic) domain, are indicated. CR indicates the connecting region that comprises the site of proteolytic activation.
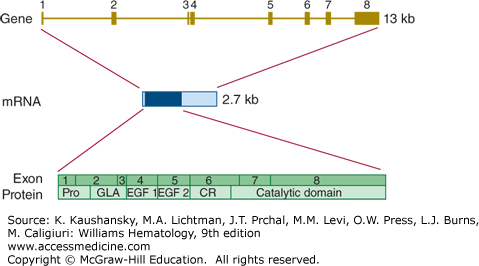
Inherited factor VII deficiency is a rare autosomal recessive disorder that affects approximately one in 500,000 newborns.30 Factor VII deficiency is the most common of the inherited rare bleeding disorders, although the reported prevalences vary between countries. Homozygotes and compound heterozygotes develop a hemorrhagic diathesis that may vary from mild to severe.
The human gene mutation database (http://www.hgmd.org) lists 258 mutations in the factor VII gene. The majority of these are missense mutations, but splicing and regulatory mutations also occur. Small deletions account for almost 10 percent of the documented mutations. Other gross gene abnormalities appear to be uncommon.
The factor VII gene harbors many common polymorphisms of which three are notable: Arg353Gln in the catalytic domain, a 10-bp insertion in the promotor region, and a variable number of 37 bp repeats in intron 7.43 The minor alleles of these polymorphisms are associated with decreased levels of factor VII and explain up to 30 percent of the variation in activated factor VII levels. Furthermore, the minor alleles have been claimed to lower the risk of myocardial infarction. However, this finding has not led to routine genotyping in the management of this disorder. The relationship between factor VII levels, factor VII polymorphisms, and venous thrombosis has not been established with certainty.
Factor IX was originally reported in 1952 as “Christmas factor,” named after one of the first identified hemophilia B patients.34,44 Factor IX is synthesized in the liver and circulates in plasma as a single-chain zymogen of 415 amino acids (Mr ≈55,000) at a concentration of 90 nM with a half-life of 18 to 24 hours (see Table 113–1).
Factor IX consists of a Gla domain, two EGF-like domains, a 35-residue activation peptide, and the serine protease domain (see Fig. 113–1). The Gla domain contains 12 Gla residues, of which the 11th and 12th Gla (Glu36 and Glu40) are not evolutionary conserved in other vitamin K–dependent proteins and are not essential for normal factor IX function.45
Factor IX comprises several posttranslational modifications that are not only important for its structure and function, but are also involved in the plasma clearance and distribution of factor IX.35 Factor IX is sulfated at Tyr155 and phosphorylated at Ser158 in the activation peptide. Hydroxylation of Asp64 in EGF 1 mediates calcium binding, and while only approximately 40 percent of total plasma factor IX carries this modification, complete absence because of a point mutation at this position dramatically reduces factor IX activity resulting in hemophilia B.46,47 An O-linked fucose (Ser61) and glucose (Ser63) are found in the EGF 1 domain, in addition to several O-linked glycans in the activation peptide (Thr159, Thr169, Thr172, and Thr179). Further modification of the activation peptide includes N-linked glycosylation of Asn residues 157 and 167, which modulates the circulating levels of factor IX.48,49,50
Factor IX binds with high affinity to the extracellular matrix component collagen IV via residue Lys5 in the Gla domain.51,52 Although factor IX variants incapable of collagen IV binding exhibit a greater recovery, collagen IV association generates an extravascular reservoir of factor IX that enables prolonged action of factor IX at a hemostatic relevant region.
Limited proteolysis of factor IX at both Arg145 and Arg180 by either the tissue factor–factor VIIa complex or factor XIa results in the release of the activation peptide and generation of factor IXa (see Fig. 113–1). Cleavage at Arg180 generates factor IXaα, which displays catalytic activity toward synthetic substrates only, whereas fully active factor IXaβ is formed following cleavage at Arg145.53,54
Factor IXa is a two-chain serine protease (Mr ≈45,000) that is composed of a light chain of 145 residues (Mr ≈17,000) and the catalytic heavy chain of 235 residues (Mr ≈28,000), which are covalently linked via a disulfide bond.
Factor IXa has a low catalytic efficiency as a result of impaired access of substrates to the active site that results from steric and electrostatic repulsion.55 Reversible interaction with the cofactor VIIIa on anionic membranes and subsequent factor X binding leads to rearrangement of the regions surrounding the active site and proteolytic factor X activation.
The primary plasma inhibitor of factor IXa is the serpin anti-thrombin, and this inhibition is enhanced by heparin (see Table 113–4), which induces a conformational change in antithrombin that is required for simultaneous active site and exosite interactions with factor IXa.56
The gene encoding factor IX (F9) is located on chromosome Xq27.1 and covers nearly 25 kb.57 It is divided into eight exons from which a mature mRNA molecule is transcribed with an ultimate length of 2802 bases (Fig. 113–8).
Figure 113–8.
Relationship of gene structure to protein structure in factor IX. The exons, introns, mRNA, and protein structure are as indicated. The mRNA is 2.8 kb with a small 5′ untranslated region and a relatively large 3′ untranslated region (light blue). In the protein, Pro indicates the prepro leader sequence, GLA indicates the γ-carboxy glutamic acid (Gla) domain, and epidermal growth factor (EGF)-1 and -2, as well as the serine protease (catalytic) domain are indicated. AP indicates the activation peptide that is released after cleavage of two bonds.
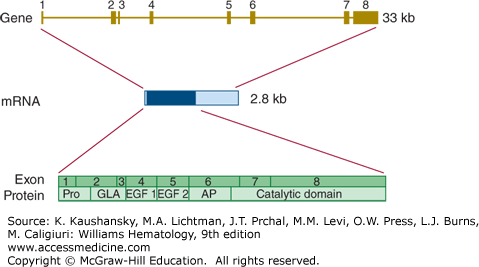
A defect or deficiency in factor IX leads to hemophilia B. Chapter 123 discusses the prevalence, clinical characteristics, and molecular genetics of hemophilia B in detail.
Conversely, increased levels of factor IX are a strong risk factor for venous thrombosis.58 This is in agreement with a rare gain of function mutation (Arg335Leu; factor IX Padua), which renders the protein hyperfunctional and is associated with familial early-onset thrombophilia.59
Factor X was originally reported in the late 1950s as the “Stuart-Prower factor,” named after the first two identified factor X–deficient patients.60,61,62 Factor X is primarily synthesized in the liver and circulates in plasma as a two-chain zymogen of 445 amino acids (Mr ≈59,000) at a concentration of 170 nM with a half-life of 34 to 40 hours (see Table 113–1).
Factor X is synthesized as a single-chain precursor and during intracellular processing, the three-amino acid peptide Arg140-Lys141-Arg142 is excised. The resulting two-chain zymogen consists of a light chain (Mr ≈16,000), comprising the Gla domain with 11 Gla residues and the EGF domains, which is linked via a disulfide bond to the heavy chain (Mr ≈42,000) that consists of a 52-residue activation peptide and the serine protease domain (see Fig. 113–1).
Hydroxylation of Asp63 mediates calcium binding to the EGF 1 domain and orients the Gla domain, which is essential for factor X clotting activity.35 N-linked glycosylation of the activation peptide residues Asn181 and Asn191 has been implicated in prolonging the factor X half-life.63 Further posttranslational modification of factor X consists of O-linked glycosylation at Thr159 and Thr171 in the activation peptide and Thr443 in the serine protease domain. There is some evidence that glycosylation of the human factor X activation peptide may also contribute to substrate recognition by the intrinsic or extrinsic factor X-activating complex.64,65
Factor X is proteolytically activated by either the factor VIIIa–factor IXa (“intrinsic tenase”) or the tissue factor–factor VIIa (“extrinsic tenase”) enzyme complexes following cleavage at Arg194 in the heavy chain (see Fig. 113–1). This results in the release of the activation peptide and generation of factor Xa, also known as factor Xaα. A snake venom protease from Russell’s viper venom (RVV-X) is capable of generating factor Xa in a similar manner.
Factor Xa consists of the Gla and EGF domains-comprising light chain (Mr ≈16,000) and the catalytic heavy chain (Mr ≈29,000) that are covalently linked via a disulfide bond. Factor Xa reversibly associates with its cofactor factor Va on an anionic membrane surface in the presence of calcium ions to form prothrombinase, the physiologic activator of prothrombin. Factor Xa is also involved in the proteolytic activation of factors V, VII, and VIII.66,67,68
Similar to thrombin, factor Xa plays a role in other biologic and pathophysiologic processes that are not directly related to coagulation. Factor Xa triggers intracellular signaling via activation of PAR1 and/or PAR2. Factor Xa cleaves PAR2 by itself as well as in complex with tissue factor–factor VIIa. These direct cellular effects of factor Xa contribute to wound healing, tissue remodeling, inflammation, angiogenesis, and atherosclerosis, among others.26,69
Further autocatalytic cleavage at Arg429 near the C-terminus of the factor Xa heavy chain leads to release of a 19-residue peptide, yielding the enzymatically active factor Xaβ.70,71,72 Plasmin-mediated cleavage of factor Xa at adjacent C-terminal Arg or Lys residues also results in the generation of factor Xaβ and factor Xaβ derivatives.73,74 While the coagulation activity is eliminated in the factor Xaβ derivatives, they are capable of interacting with the zymogen plasminogen and enhance its tissue plasminogen activator-mediated conversion to plasmin, thereby promoting fibrinolysis.75
A primary plasma inhibitor of factor Xa is the serpin anti-thrombin, and this inhibition is enhanced by heparin (see Table 113–4), which induces a conformational change in antithrombin that is required for simultaneous active site and exosite interactions with factor Xa.76 Another potent factor Xa inhibitor is TFPI, which inhibits both the ternary tissue factor–factor VIIa–factor Xa complex as well as free factor Xa, for which protein S functions as a cofactor.77,78 Free factor Xa is also inhibited by the protein Z/protein Z–dependent protease inhibitor (ZPI) complex on membranes.79
Low-molecular-weight heparin and synthetic derivatives (e.g.fondaparinux) are clinically used as anticoagulants to enhance factor Xa inhibition by antithrombin specifically. The target-specific oral anticoagulants rivaroxaban, apixaban, edoxaban, and analogues directly inhibit both free factor Xa and prothrombinase complex-assembled factor Xa with high specificity through a high-affinity, reversible interaction with the factor Xa active site.80,81,82,83
The gene encoding factor X (F10) is located on chromosome 13q34 and spans almost 27 kb.84 The 8 exons in the factor X gene give rise to a mature mRNA of 1560 bases (Fig. 113–9). There are no common alternative splice variants with known biology.
Figure 113–9.
Relationship of gene structure to protein structure in factor X. The exons, introns, mRNA, and protein structure are as indicated. The mRNA is 1.5 kb with a relatively large 5′ untranslated region and a small 3′ untranslated region. In the protein, Pro indicates the prepro leader sequence, GLA indicates the γ-carboxy glutamic acid (Gla) domain, and epidermal growth factor (EGF)-1 and -2, as well as the serine protease (catalytic) domain, are indicated. AP indicates the activation peptide. Before secretion, cleavage in this domain processes factor X to the two-chain mature zymogen. A second cleavage releases the activation peptide and generates factor Xa activity.
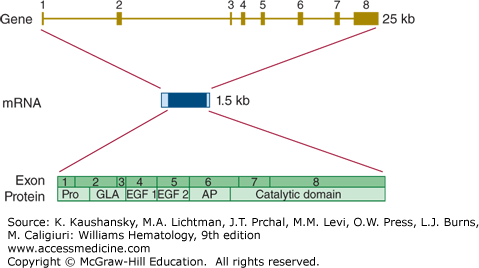
Loss of function mutations in the factor X gene lead to a rare bleeding disorder with a recessive mode of inheritance. Factor X deficiency occurs in approximately one in every 1,000,000 newborns. Most cases of documented factor X deficiency experience serious bleeding problems. In fact, factor X deficiency may be the most severe of the rare congenital bleeding disorders.30 Well over 100 mutations have been documented in cases with factor X deficiency (http://www.hgmd.org). The majority of these mutations are missense and nonsense mutations.
Gain-of-function mutations in factor X could potentially increase thrombotic risk, but such mutations have not been documented. There is uncertainty about whether common gene variations influence the level of factor X in plasma.85
Protein C, which plays a central role in the anticoagulant pathway, was discovered in 1960, and being the third protein peak (“peak C”) observed in a vitamin K–dependent plasma protein purification, it was named protein C.86,87 Protein C is synthesized in the liver and circulates in plasma as a two-chain zymogen of 417 amino acids (Mr ≈62,000) at a concentration of 65 nM with a half-life of 6 to 8 hours (see Table 113–1).
Protein C is synthesized as a single-chain precursor and during intracellular processing amino acids Lys146-Arg147 are excised. The resulting two-chain zymogen consists of a light chain (Mr ≈21,000) comprising the Gla domain with nine Gla residues and the EGF domains, which is linked via a disulfide bond to the heavy chain (Mr ≈41,000) that consists of the 12-residue activation peptide and the serine protease domain (see Fig. 113–1).
In addition to γ-carboxylation, protein C is hydroxylated at Asp71 in the EGF-1 domain, which coordinates calcium binding.35 N-linked glycosylation of Asn97 in EGF-1 and Asn248, Asn313, and Asn329 in the serine protease domain are important for efficient protein secretion, proteolytic processing of Lys146-Arg147, and proteolytic activation.88,89,90 Some of the total plasma protein C is not glycosylated at either Asn329 (β-protein C) or at both Asn329 and Asn248 (γ-protein C), of which the impact on protein function remains unclear.91
Protein C is proteolytically activated by α-thrombin in complex with the endothelial cell surface protein thrombomodulin following cleavage at Arg169 (see Fig. 113–1). The activation peptide is released and the mature serine protease activated protein C (APC) is formed. Activation of protein C is enhanced by its localization on the endothelial surface through association with the endothelial cell protein C receptor (EPCR).92 Several snake venom proteases (RVV-X and Protac) are also capable of activating protein C.
APC consists of the disulfide-linked light chain comprising the Gla and EGF domains (Mr ≈21,000) and the catalytic heavy chain (Mr ≈32,000). In complex with its cofactor protein S, APC proteolytically inactivates factors Va and VIIIa in a calcium- and membrane-dependent manner. Intact factor V has been reported to function as a cofactor for the inactivation of factor VIIIa in the presence of protein S.93
Downregulation of thrombin formation through inactivation of these cofactors seems to occur preferentially on the endothelial cell surface as opposed to that of platelets,94 where it prevents coagulation and potential thrombosis. However, protein C activation is also accelerated by platelet factor 4 (PF4), which is secreted by activated platelets. Upon interaction with the Gla domain of protein C, PF4 modifies the conformation of protein C, thereby enhancing its affinity for the thrombomodulin-thrombin complex.95 This ensures APC generation in close proximity of the injury site where platelets are activated, which serves to impede dissemination of coagulation.
APC also plays a major role in the cytoprotective pathway to prevent vascular damage and stress.96 These activities include antiapoptotic activity, antiinflammatory activity, alterations of gene-expression profiles, and endothelial barrier stabilization. Most of these functions require binding to EPCR and PAR1 cleavage.
APC is primarily inhibited by the heparin-dependent serpin protein C inhibitor and by plasminogen activator inhibitor-1 (PAI-1). Because PAI-1 is the major inhibitor of tissue plasminogen activator, inhibition through complex formation with APC contributes to enhanced fibrinolysis. Chapter 114 discusses these and other factors that attenuate the anticoagulant activity of APC.
The protein C gene (PROC) is located on chromosome 2q14.3 and spans almost 11 kb.97 The gene is divided into nine exons and the mature mRNA has a length of 1790 bases (Fig. 113–10). There are no alternative mRNA species with known biology.
Figure 113–10.
Relationship of gene structure to protein structure in protein C. The exons, introns, mRNA, and protein structure are as indicated. The mRNA is 1.8 kb with a small 5′ untranslated region coded for by exon 1 and a relatively small 3′ untranslated region (light blue). In the protein, Pro indicates the prepro leader sequence, GLA indicates the γ-carboxy glutamic acid (Gla) domain, and epidermal growth factor (EGF)-1 and -2, as well as the serine protease (catalytic) domain, are indicated. AP indicates the activation peptide. Before secretion, cleavage in this domain processes protein C to the two-chain mature zymogen. A second cleavage releases the activation peptide and generates activated protein C.
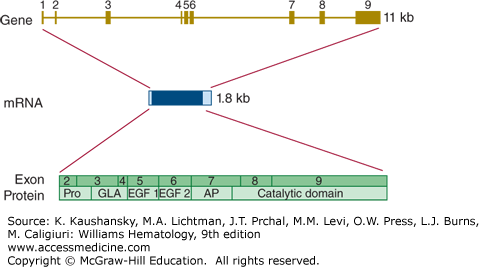
Loss-of-function mutations cause protein C deficiency. In homozygous or compound heterozygous form this leads to life-threatening purpura fulminans at birth which, if left untreated, is fatal.98 In cases where there is still some protein C activity detectable, symptoms may be much milder.
Heterozygous protein C deficiency increases the risk of venous thrombosis. This is true for most deficiencies of natural anticoagulants and sets them apart from rare bleeding disorders where heterozygosity for loss of function mutations is mostly asymptomatic. The risk for venous thrombosis is increased approximately 10-fold in heterozygotes for protein C deficiency, albeit that the risk estimates vary considerably between studies.99 Family studies in particular suggest a high risk, whereas case-control studies may show markedly lower estimates.100
Heterozygous protein C deficiency can be categorized as type I or type II. In type I deficiency, antigen levels are approximately 50 percent of normal, whereas in type II deficiency, antigen levels are (near) normal but activity levels are decreased by 50 percent.
The genetic basis of protein C deficiency, consistent with what is observed in general for congenital loss of function disorders, is heterogeneous. In line with this, more than 300 mutations have been documented and are tracked in the human gene mutation database (http://www.hgmd.org). Two-thirds of these documented mutations are missense or nonsense.
Several common polymorphisms, in particular in the promotor region of the protein C gene, are known to have a small but measurable effect on plasma protein C levels. Alleles of these polymorphisms that are associated with lower protein C levels are also associated with an increased thrombotic risk, albeit that the effect is small.101 Therefore, it is not surprising that measurement of these polymorphisms have not found any clinical application.
THE PROCOAGULANT COFACTORS V AND VIII
Factors V and VIII both function as cofactors in coagulation and dramatically enhance the catalytic rate of their macromolecular enzyme complexes, resulting in the generation of thrombin and factor Xa, respectively. Apart from their functional equivalence, they also share similar gene structures, amino acid sequences, and protein domain structures, which is not surprising considering that factors V and VIII are assumed to descend from the common ancestral A1-A2-A3 domain-containing copper-binding plasma protein ceruloplasmin through a gene duplication event.102 After acquiring C-type domains as well as the central B domain, a second gene duplication ultimately separated the ancestral genes of factors V and VIII.
Factors V and VIII undergo similar mechanisms of intracellular processing in the endoplasmic reticulum (ER) and Golgi apparatus. Trafficking through this early secretory pathway involves interaction of factors V and VIII with a receptor complex that consists of the mannose-binding lectin-1 gene product LMAN1 (also called ER-Golgi intermediate compartment (ERGIC)-53) and multiple coagulation deficiency protein 2 (MCFD2).103 Defects or deficiencies in one of the two subunits of the receptor complex can result in a combined deficiency of factors V and VIII (Chap. 124).
In 1943, Norwegian physician Paul Owren discovered the fifth coagulation factor thus far known and named it factor V.104,105,106 Factor V is synthesized in the liver and circulates in plasma as a large single-chain procofactor of 2196 amino acids (Mr ≈330,000) at a concentration of 20 nM with a half-life of 12 to 36 hours (see Table 113–1).
Approximately 20 percent of the total factor V in blood is stored in the α-granules of platelets. Although it was originally thought that megakaryocytes synthesize factor V, studies in humans indicate that platelet factor V originates from plasma through endocytic uptake.107,108,109 Platelet factor V is modified intracellularly such that it is functionally unique compared to its plasma-derived counterpart. It is partially activated, more resistant to inactivation by APC, and has several different posttranslational modifications.110
Platelet factor V is associated with the large multimeric protein multimerin.111 Multimerin has a massive repeating structure, with some of the multimers having molecular weights of several million daltons. Although the function of this platelet factor V-specific multimeric chaperon protein is similar to that of VWF, the multimeric chaperon protein of factor VIII in plasma, multimerin and VWF share no structural homology.
Following platelet activation, platelet factor V becomes available at the site of injury and can reach local concentrations that exceed the factor V plasma concentration by more than 100-fold.112 Interestingly, the origin of factor V in mouse platelets differs from humans in that it is synthesized in megakaryocytes and stored into the α-granules before platelets are released from the marrow.113,114
Factor V has an A1-A2-B-A3-C1-C2 domain structure (Fig. 113–11). The three A-type domains share significant homology with those of ancestral ceruloplasmin as well as with the factor VIII A domains (approximately 50 percent sequence identity). The two C-type domains belong to the family of discoidin domains, which are generally involved in cell adhesion, and share approximately 55 percent sequence identity with the factor VIII C domains. The C domains mediate binding to the anionic phospholipid surface, thereby localizing factor V to the site of injury and facilitating interaction with factor Xa and prothrombin.115,116,117,118 In contrast, the large central B domain of factor V shows weak homology to the factor VIII B domain or to any other known protein domain. However, this domain comprises so-called basic and acidic regions that are highly conserved throughout evolution and serve to negatively regulate factor V function and prevent activity of the procofactor.119,120
Figure 113–11.
The domain structure of factor V. Schematic A1-A2-B-A3-C1-C2 domain representation of factor V. Thrombin cleavage sites (Arg709, Arg1018, Arg1545) are indicated by green arrows, and activated protein C (APC) cleavage sites (Arg306, Arg506) by red arrows. The blue and red boxes in the B domain represent the basic and acidic region, respectively, that are highly conserved throughout evolution and serve to negatively regulate factor V function and prevent activity of the procofactor V.

Factor V undergoes extensive posttranslational modifications, including sulfation, phosphorylation, and N-linked glycosylation.35,121 Sulfation at sites in the A2 and B domain are involved in the thrombin-mediated proteolytic activation of factor V.122 Phosphorylation at Ser692 in the A2 domain enhances the APC-dependent inactivation of the cofactor Va.123 N-linked glycosylation occurs throughout the whole protein; however, the majority of carbohydrates are linked to Asn residues within the B domain and play a role in the LMAN1-MCDF2 receptor complex-mediated trafficking of factor V from the ER to the Golgi in the early secretory pathway.103 Partial glycosylation at Asn2181 in the C2 domain of factor V results in a lower binding affinity for negatively charged membranes of the glycosylated form, thereby reducing the factor V procoagulant activity, particularly at low phospholipid concentrations.124,125 Furthermore, factor V comprises several disulfide bonds that are important for the three-dimensional structure of the A and C domains.121
Sequential proteolytic cleavage of the procofactor factor V at Arg709, Arg1018, and Arg1545 in the B domain results in release of the inhibitory constraints exerted by the B domain and in the generation of the heterodimeric cofactor Va (see Fig. 113–11).126 Maximal cofactor activity correlates with cleavage at Arg1545, which is consistent with the observation that a snake venom protease from RVV-V, which cleaves only at Arg1545, results in full activation. Thrombin has generally been recognized to be the principal activator of factor V. However, recent findings suggest that in the initiation phase of coagulation factor V is primarily activated by factor Xa.127 Factor Xa initially cleaves factor V at Arg1018, followed by proteolysis at Arg709 and Arg1545.128
Factor Va is composed of a heavy chain (Mr ≈105,000) comprising the A1-A2 domains and the A3-C1-C2 light chain (Mr ≈74,000), which are noncovalently associated via calcium ions. Factor Va is a nonenzymatic cofactor within the prothrombinase complex that greatly accelerates the ability of factor Xa to rapidly convert prothrombin to thrombin.129 APC catalyzes the inactivation of factor Va by cleavage at the main sites Arg306 and Arg506, upon which the cleaved A2 fragment dissociates and factor Va can no longer associate with factor Xa.130 A common Arg506Gln mutation in factor V leads to resistance to inactivation by APC (factor V Leiden) and is associated with an increased risk of venous thromboembolism (Chap. 133).131
Both factor V and an alternatively spliced isoform of factor V (factor V-short), which lacks the major part of the B domain (residues 756 to 1458) and normally circulates in low abundance, interact with full-length TFPI (TFPIα), most likely through the acidic B domain region.132,133 The linkage of factor V and TFPIα is considered to attenuate the bleeding phenotype in factor V–deficient patients, as the low TFPIα levels in these patients allow the residual platelet factor V to be sufficient for coagulation.132,134 Conversely, increased factor V–short expression caused by an A2440G mutation in the factor V gene leads to a dramatic increase in plasma TFPIα, resulting in a bleeding disorder.133
The gene for factor V (F5) is located on chromosome 1q23. It is located very close to the genes for the selectin family of leukocyte adhesion molecules. The factor V gene spans approximately 70 kb and consists of 25 exons (Fig. 113–12). The gene structure is very similar to that of the factor VIII gene, with exon–intron boundaries occurring at exactly the same location in 21 out of 24 cases.135
Figure 113–12.
Relationship of gene structure to protein structure in factor V. The exons, introns, mRNA, and protein structure are as indicated. The mRNA is 7 kb with some 5′ and 3′ untranslated sequences (light blue). In the protein, P indicates the propeptide leader sequence, and the A1-A2-B-A3-C1-C2 domains are indicated.
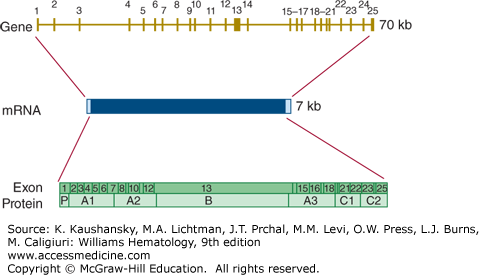
Homozygosity or compound heterozygosity for loss-of-function mutations in the factor V gene lead to a bleeding disorder (termed parahemophilia or Owren parahemophilia).136 At the time of writing, 152 mutations in the factor V gene have been collected in the human gene mutation database (www.hgmd.org).
Gain-of-function mutations in the factor V gene increase the risk of thrombosis. This is particularly the case for venous thrombosis and not so much for arterial thrombosis. In whites, the most common gain-of-function mutation in the factor V gene is factor V Leiden (Arg506Gln), which leads to a plasma abnormality known as APC resistance (Chap. 133).137,138
Factor VIII (antihemophilic factor) was first discovered in 1937, but it was not until 1979 that its purification by Tuddenham and coworkers led to the molecular identification of the protein.139,140 Factor VIII is synthesized as a single-chain preprocofactor of 2351 amino acids and, subsequent to intracellular processing, is secreted as a series of metal ion-linked heterodimers due to proteolysis at the A3-B junction and differential processing in the central B domain (Fig. 113–13). The mature factor VIII procofactor comprises 2332 amino acids (Mr ≈300,000) and circulates in a high-affinity complex with its carrier protein VWF at a concentration of approximately 0.7 nM and a circulatory half-life of 8 to 12 hours (see Table 113–1). Complex formation with VWF protects factor VIII from proteolytic degradation, premature ligand binding, and rapid clearance from the circulation.
Figure 113–13.
The domain structure of factor VIII. Schematic A1-a1-A2-a2-B-a3-A3-C1-C2 domain representation of factor VIII. The acidic regions denoted by a1, a2, and a3 are indicated, thrombin cleavage sites (Arg372, Arg740, Arg1689) are indicated by green arrows, and activated protein C (APC) cleavage sites (Arg336, Arg562) by red arrows. The variably sized B domain as a result of differential proteolytic processing is indicated.

The primary source of factor VIII is the liver,141,142 but extrahepatic synthesis of factor VIII also occurs.143,144 While contradictory evidence exists on the cellular origin of both hepatic and extrahepatic factor VIII synthesis, recent studies in mice support that endothelial cells from many tissues and vascular beds synthesize factor VIII, with a large contribution from hepatic sinusoidal endothelial cells.145,146,147 This is consistent with observations on factor VIII expression in human endothelial cells from the liver and lung.148,149
Factor VIII is less-efficiently secreted from the cell as compared to factor V, because it interacts with the ER-chaperon proteins calnexin and calreticulin, whereas factor V interacts with calreticulin only.150 Both chaperons preferentially interact with GPs comprising mono-glucosylated N-linked oligosaccharides and promote correct folding of proteins that enter the secretory pathway and target misfolded proteins for degradation. Factor VIII, but not factor V, also interacts with the ER-chaperon immunoglobulin-binding protein (BiP/GRP78), which appears to enhance the stability of factor VIII, but also retards its secretion.151 Factor VIII trafficking from the ER to the Golgi is mediated via the LMAN1-MCDF2 receptor complex, similar to factor V.103
Several clearance receptors are responsible for actively removing factor VIII from the circulation, which include the low-density lipoprotein (LDL) receptor-related protein 1 (LRP1), the LDL receptor, and receptors that specifically interact with carbohydrate structures on factor VIII.152,153,154,155,156
The A1-A2-B-A3-C1-C2 domain structure of factor VIII shares significant homology with factor V except in the B domain region (see Fig. 113–13). In contrast to factor V, the factor VIII B domain is dispensable for procoagulant activity. The mature factor VIII procofactor comprises a variably sized heavy chain (A1-A2-B; Mr ≈200,000 to 90,000 depending on the extent of proteolysis) and a light chain (A3-C1-C2; Mr ≈80,000). The C-terminal regions of the A1 and A2 domains and the N-terminal portion of the A3 domain contain short segments of 30 to 40 negatively charged residues known as the a1, a2, and a3 regions. Interaction with VWF is facilitated by the a3 region and C1 domain.157,158 The C domains mediate binding to the anionic phospholipid surface, thereby localizing factor VIII to the site of injury and facilitating interaction with factor IXa and factor X.159,160,161
Factor VIII is heavily glycosylated and the majority of the N-linked glycosylation sites are found in the B domain, which mediate interaction with the chaperons calnexin and calreticulin and, in part, with the LMAN1–MCDF2 receptor complex.103,150,162 Sulfation of tyrosine residues is required for optimal activation by thrombin, maximal activity in complex with factor IXa, and maximal affinity of factor VIIIa for VWF.35,163 Factor VIII comprises two phosphorylation sites that are located in the A1 (Thr351) and B (Ser1657) domains.
Thrombin and factor Xa are the principal activators of the procofactor VIII and generate the cofactor VIIIa through sequential proteolysis at Arg740, Arg372, and Arg1689.126,164,165,166 The heterotrimeric factor VIIIa is composed of the A1 (Mr ≈50,000), A2 (Mr ≈43,000), and the A3-C1-C2 light chain (Mr ≈73,000) subunits (see Fig. 113–13). The A1 and A3-C1-C2 subunits are noncovalently linked through calcium ions, whereas A2 is associated with weak affinity primarily by electrostatic interactions.167,168 Once activated, factor VIIIa functions as a cofactor for factor IXa in the phospholipid-dependent conversion of factor X to factor Xa. The rapid and spontaneous loss of factor VIIIa cofactor activity is attributed to A2 domain dissociation from the heterotrimer.167,168 Additional proteolysis by APC, factor Xa, or factor IXa also results in the downregulation of factor VIIIa cofactor activity.169
The factor VIII encoding gene (F8) is situated at chromosome Xq28. The factor VIII gene contains 26 exons (Fig. 113–14), one more than factor V, because exon 5 of factor V corresponds to exons 5 and 6 of the factor VIII gene.170 In addition, the gene for factor VIII is much larger than that of factor V, spanning approximately 190 kb. This is largely because six of the introns in the factor VIII gene are much larger than the corresponding F5 introns. The mRNA for factor VIII is also much larger than the factor V mRNA because of a 1.8 kb 3′-untranslated region.
Figure 113–14.
Relationship of gene structure to protein structure in factor VIII. The exons, introns, mRNA, and protein structure are as indicated. The mRNA is 9 kb with some 5′ untranslated sequence and a large 3′ untranslated region (light blue). In the protein, P indicates the propeptide leader sequence, and the A1-A2-B-A3-C1-C2 domains are indicated.
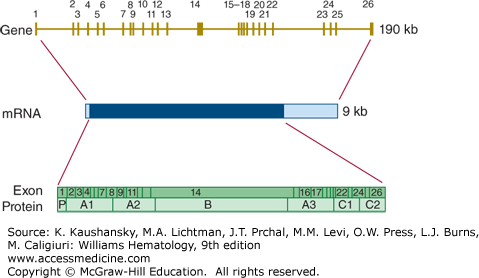
A defect or deficiency in factor VIII leads to hemophilia A. Chapter 123 discusses the prevalence, clinical characteristics, and molecular genetics of hemophilia A in detail.
High levels of factor VIII are a common and strong risk factor for venous thrombosis. It has been suspected that certain genetic variations in the factor VIII gene might play a role in determining the level of factor VIII; however, such variations have not been identified.171 The ABO blood group does play a role in determining the level of factor VIII, but probably indirectly through an effect on the level of VWF.172,173
THE SOLUBLE COFACTORS PROTEIN S AND VON WILLEBRAND FACTOR
Protein S, which is named after the city (Seattle) where it was discovered by the group of Earl Davie in 1977, is a vitamin K–dependent single-chain GP of 635 amino acids (Mr ≈75,000) that circulates with a plasma half-life of 42 hours (see Table 113–1). Part of the total protein S pool circulates in a free form at a concentration of 150 nM, whereas the majority (approximately 60 percent; 200 nM) circulates bound to the complement regulatory protein C4b–binding protein (C4BP). Protein S is primarily synthesized in the liver by hepatocytes, in addition to endothelial cells, megakaryocytes, testicular Leydig cells, and osteoblasts.174,175,176,177,178
The protein structure of protein S differs from the other vitamin K–dependent proteins as it lacks a serine protease domain and, consequently, is not capable of catalytic activity. Protein S is composed of a Gla domain comprising 11 Gla residues, a thrombin-sensitive region (TSR), four EGF domains, and a C-terminal sex hormone–binding globulin (SHBG)-like region that consists of two laminin G-type domains (Fig. 113–15). The SHBG-like domain is involved in the interaction with the β-subunit of C4BP.
Figure 113–15.
Protein S, factor XI, factor XII, and tissue factor pathway inhibitor (TFPI). Schematic of protein S, factor XI, factor XII, and TFPIα. Each circle represents an amino acid. For protein S: the prepro leader sequence comprising the signal peptide as well as the propeptide is indicated, the γ-carboxy glutamic acid (Gla) domain is indicated with the Gla residues in blue, TSR represents the thrombin-sensitive region, the four epidermal growth factor (EGF) domains are indicated, and SHBG represents the sex hormone–binding globulin-like region. For factor XI: the four apple domains are indicated, and the serine protease (catalytic) domain is shown. Cys321 in the apple 4 domain that forms a disulfide link with Cys321 in the other factor XI subunit, thereby mediating dimerization, is indicated in yellow. For factor XII: types I and II represent the fibronectin types I and II domains, the two EGF-like domains are indicated, the kringle domain is indicated, Pro indicates the proline-rich region, and the serine protease (catalytic) domain is indicated. For TFPIα: the three Kunitz domains are indicated and the C-terminal sequence of basic residues is indicated in light blue. Factors XI and XII have homologous serine protease (“catalytic”) domains (shown in light red), in which the active site His, Asp, and Ser residues are indicated in dark red. Cleavages that convert the zymogens factor XI and factor XII to an active enzyme are indicated by the red arrows. Cysteine residues that form a disulfide bridge (black line) are indicated in green.
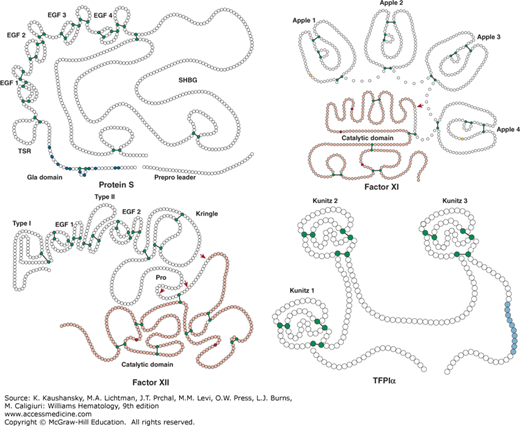
Apart from γ-carboxylation of Glu residues, protein S is posttranslationally modified via N-glycosylation in the second laminin G-type domain of the SHBG-like region (Asn458, Asn468, Asn489). β-Hydroxylation of Asp95 or Asn residues (Asn136, Asn178, Asn217) in each EGF domain allows for calcium binding that orients the four EGF domains relative to each other.35
Free protein S serves as a cofactor for APC in the proteolytic inactivation of factors Va and VIIIa.179,180 Interaction of protein S with APC on a negatively charged membrane surface alters the location of the APC active site relative to factor Va,181 which accounts for the selective protein S-dependent rate enhancement of APC cleavage at Arg306 in factor Va.182 C4BP-bound protein S also exerts a similar stimulatory effect on Arg306 cleavage, albeit to lower extent, whereas it inhibits the initial APC-mediated factor Va cleavage at Arg506, resulting in an overall inhibition of factor Va inactivation.183 Cleavage of the TSR by thrombin and/or factor Xa results in a loss of APC-cofactor activity.184 Protein S also functions as a cofactor for TFPIα in the inhibition of factor Xa, which is mediated by the SHBG-like region in protein S.77,185
Protein S has been implied to play a role in phagocytosis of apoptotic cells, cell survival, activation of innate immunity, vessel integrity and angiogenesis, and local invasion and metastasis through interaction with a family of protein tyrosine kinase receptors referred to as Tyro-3, Axl and Mer (TAM) receptors.186,187
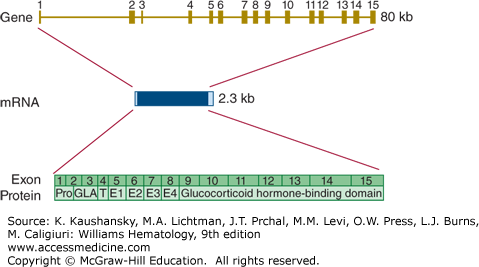
The gene encoding protein S (PROS1) is located on the long arm of chromosome 3 (3q11.1), very close to the centromere. A highly homologous protein S pseudogene (PROSP) is located on the other side of the centromere. This pseudogene is inactive, as it is not transcribed into mRNA.188 The active protein S gene encompasses 15 exons and covers a little more than 100 kb (Fig. 113–16). The mRNA sequence consists of 3560 bases. Several alternative transcripts have been identified, but none of these have known biology.
Figure 113–16.
Relationship of gene structure to protein structure in protein S. The exons, introns, mRNA, and protein structure are as indicated. The mRNA is 2.3 kb with small 5′ and 3′ untranslated regions (light blue). In the protein, Pro indicates the prepro leader sequence, GLA indicates the γ-carboxy glutamic acid (Gla) domain, T indicates the thrombin-sensitive region; E indicates the epidermal growth factor (EGF)-like domains, and the glucocorticoid hormone-binding domain represents the sex hormone–binding globulin (SHBG)-like domain.
Loss-of-function mutations in PROS1 lead to protein S deficiency. Several cases of homozygous and compound heterozygous protein S deficiency have been described with extremely low protein S levels. These very rare cases suffer from life-threatening purpura fulminans at birth.189
Much more common are heterozygous deficiencies of protein S, which can be categorized into three types of deficiency. Type I deficiency is characterized by antigen levels that are approximately 50 percent of normal. In type II deficiency, antigen levels are (near) normal while activity levels are decreased by 50 percent. Type III deficiency is defined by a low level of free protein S. In keeping with this classification, clinical chemistry laboratories may offer a protein S activity assay, free antigen assay, or total antigen assay (or a combination thereof). These assays are not without problems and the evaluation of protein S levels is fraught with complications that need careful attention before a final diagnosis can be made.190
The genetic basis of protein S deficiency is highly heterogeneous and there are more than 200 entries in the human gene mutation database (www.hgmd.org). Most of these are missense mutations. However, protein S deficiency is often characterized by gross gene deletions, that sometimes even involve neighboring genes.191 The reason for this preponderance of gross gene abnormalities remains unknown.
It is commonly assumed that protein S deficiency increases venous thrombotic risk by 10-fold.100 This assertion is mainly based on studies in thrombophilic families. In population-based case-control studies, however, the risk increase appears to be much more modest, if present at all.192 The reason for this discrepancy between family and population-based studies remains enigmatic. The findings argue against including tests for protein S deficiency in a thrombophilia workup of venous thrombosis cases with a negative family history.
Chapter 126 discusses the structure, function, and molecular biology of VWF in detail. VWF is a large multimeric GP that is required for normal platelet adhesion to components of the vessel wall and that serves as a carrier for factor VIII. It is exclusively synthesized in megakaryocytes and endothelial cells and stored in specialized organelles in platelets and endothelial cells. Release of VWF multimers from these organelles follows upon a stimulus or via unstimulated basal secretion from endothelial cells.193 VWF multimers circulate at a concentration of 10 nM with a half-life of 8 to 12 hours (see Table 113–1). Clearance of VWF multimers is mainly mediated by macrophages from the liver and spleen.194
Large VWF multimers are cleaved by the plasma protease ADAMTS-13 (a disintegrin and metalloproteinase with thrombospondin motifs 13).195 This cleavage produces the smaller size VWF multimers that circulate in plasma. Reduced ADAMTS-13 activity is linked to various microangiopathies with increased platelet activity.
The precursor protein of VWF is composed of a 22-residue signal peptide and of a proVWF protein comprising 2791 amino acids that has 14 distinct domains in the order of D1-D2-D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK.196 Upon translocation to the ER, the signal peptide is cleaved off, and the proVWF dimerizes in a tail-to-tail fashion through cysteines in its cysteine knot (CK) domain. During transit through the Golgi apparatus, proVWF dimers multimerize in a head-to-head fashion through the formation of disulfide bonds between cysteine residues in the D3 domain. At the same time, D1 and D2 domains are cleaved off as a single fragment to form the VWF propeptide (741 amino acids), while the remaining domains comprising 2050 amino acid residues and up to 22 carbohydrate chains form mature VWF. In the trans-Golgi network, the VWF propeptide promotes mature VWF to assemble into high-molecular-weight multimers (Mr ≈500,000 to 20,000,000). These multimers subsequently aggregate into tubular structures that are packaged into α-granules in megakaryocytes and into Weibel-Palade bodies in endothelial cells.
Upon exocytosis from Weibel-Palade bodies and at high shear rates, multimeric VWF unrolls from a globular to a filamental conformation (often called VWF strings
Related posts:
 Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


