Methodology of Clinical Trials
In this chapter, we discuss the design, conduct, and analysis of oncology clinical trials, pointing out particular areas of interest to radiation oncology and reviewing some recent ideas in clinical trials and related research studies. This chapter provides a brief and essentially nontechnical sketch of the main concepts and current research areas, and we refer the reader to comprehensive texts on clinical trial conduct in oncology for further details. Excellent recent texts, such as Handbook of Statistics in Clinical Oncology, Clinical Trials in Oncology, and Oncology Clinical Trials: Successful Design, Conduct and Analysis provide the fundamentals, as well as up-to-date discussion of new challenges and active research in statistical methods for oncology clinical trials.1–3
Clinical trials enable physicians to advance medical care in a safe, scientific, and ethical manner. Formally defined, clinical trials are a set of procedures in medical research conducted to allow safety and efficacy data to be collected for health interventions.4 A more detailed definition for our purposes would describe a clinical trial as a prospective study that includes an active intervention, carried out in a well-defined patient cohort and producing interpretable information about the action of the intervention.5 Although in some ways similar to a well-designed laboratory experiment, the involvement of living human subjects demands adherence to strict ethical principles while also adding to the complexity of interpretation of the results. The past 60 years have witnessed an unprecedented appreciation of the importance of clinical trials and a consequent surge in the number of clinical trials performed. In the field of radiation oncology alone, according to a PubMed search, 376 clinical trials were published in 2010, of which 65 were phase III trials.
Over the course of less than a century, the evidence on which medicine is practiced has evolved from being entirely empirical to being a highly regulated scientific process based on vigorously designed clinical trials tightly overseen by numerous scientific and governmental agencies. Cardinal chapters in the history of clinical trial design and implementation include the following:
• 1747—James Lind’s work on the effect of citrus fruits in the prevention of scurvy among sailors in the Royal Navy
• 1863—Austin Flint’s use of a placebo group for comparison with an experimental treatment in the treatment of rheumatic fever
• 1947—Nuremberg Code, a result of the appreciation that much of the medical experimentation performed by physicians in Nazi Germany was both ethically wrong and scientifically uninterpretable
• 1948—The first double blind trial, performed by the British Medical Research Council to assess the value of streptomycin in the treatment of tuberculosis
• 1964—Declaration of Helsinki developed by the World Medical Association, ethical guidelines, which continue to be updated, for the performance of clinical trials
• 2000—Creation of the ClinicalTrials.gov Web site, a registry of clinical trials under the auspices of the National Institutes of Health (NIH)
The performance of high-quality cancer clinical trials involves the cooperation of multiple bodies, so-called stakeholders, including cancer patients and their families, physicians (who accrue the patients), their operating environment (academic institution or practice), the research team (who runs the trial on a day-to-day basis), sponsors (who oversee and fund the trial), independent monitors (to ensure the correct performance of the research team), government regulatory agencies, contract research organizations (CROs, which may carry out specific aspect of the trial such as auditing), and medical insurance companies. Modern clinical trials may require involvement of translational scientists, and clinical psychologists, as well as experts in quality of life, cost-effectiveness, and other disciplines. The complexity of clinical trials adds to the regulatory work involved. For instance, a multi-institutional federally sponsored clinical trial protocol will require the approval of at least three different research ethics oversight committees (institutional review boards [IRBs]) within the group coordinating the trial, at each institution that opens the trial to accrue patients, and at the sponsor.
Although many phase I and phase II trials are carried out by investigators within a single institution, many larger phase II and most phase III trials are generally multi-institutional. Thus, clinical trial investigator networks have emerged as essential players in the performance of large phase II and III clinical trials. The U.S. National Cancer Institute–sponsored Cancer Cooperative Group Program is an example,6 as are similar groups such as the European Organisation for Research and Treatment of Cancer (EORTC). One cooperative group, the Radiation Therapy Oncology Group (RTOG), founded by Simon Kramer in 1968, is dedicated to trials involving radiation therapy, has activated 460 protocols, and has accrued approximately 90,000 patients to its trials. Early studies sought to answer questions regarding radiation dose and fractionation. As cancer therapy became multimodal, the group addressed questions relating to combining systemic chemotherapy with radiation therapy. Most recent trials seek to combine targeted agents with contemporary radiation therapy techniques.6
Clinical trials are extremely expensive to perform, with costs continuing to rise as a result of both increased regulatory oversight and greater trial complexity. It has been estimated that implementation of the European Union’s Clinical Trials Directive (laws and regulations related to implementation of good clinical practice in the conduct of clinical trials) led to a doubling of the cost of running noncommercial cancer trials in the United Kingdom.7 Large phase III trials can cost in excess of $100 million; as a result, clinical trials are frequently financed by the pharmaceutical industry. An unfortunate consequence is that clinical trials are rarely performed on established generic drugs, where there is little commercial interest in establishing new indications. Conversely, the performance of rigorous clinical trials contributes significantly to the costs involved in the development of new pharmaceutical agents, which are subsequently reflected in the commercial pricing of the product.
TABLE 14.1 COHERENT FRAMEWORK FOR DETERMINING WHETHER CLINICAL RESEARCH IS ETHICAL

 OVERVIEW OF ETHICAL CONSIDERATIONS
OVERVIEW OF ETHICAL CONSIDERATIONS
Medical ethics are based on the principles of autonomy (the patient’s right to refuse or choose treatment), beneficence (a practitioner should act in the best interest of the patient), nonmaleficence (first, do no harm), justice (fairness and equality), dignity, and honesty. Without due diligence, physicians may infringe on these principles when encouraging patient participation in clinical trials. Are the physicians confident that the proposed treatment is beneficial and not harmful? Are the potential subjects fully aware of the implications of participation? Do all segments of the population have equivalent chance to participate and receive potentially better treatment? Despite the universal acceptance of these principles, there have been numerous examples of grossly unethical research being performed in the Western world within living memory. Documents seeking to address these issues include the Nuremberg Code, the declaration of Helsinki, and the Belmont Report. Emanuel et al.8 have listed seven requirements that provide a systematic and coherent framework for determining whether clinical research is ethical (Table 14.1).
Ethical principles themselves and the creation of guidelines are insufficient to ensure the ethical conduct of medical research. Physicians within Nazi Germany performed atrocities despite the existence of German guidelines published in 1931,9 reflecting the need for legislation. The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) brought together the regulatory authorities of Europe, Japan, and the United States and experts from the pharmaceutical industry to regulate scientific and technical aspects of pharmaceutical product registration. The ICH guidelines are legally binding in many countries (although not the United States) and are updated every few years, reflecting the increasing sophistication of the field. An example of a recent addition is the introduction of data and safety monitoring committees (DSMCs), which are independent groups of experts who monitor patient safety and treatment efficacy data in ongoing clinical trials. Another recent advance is the requirement for the registration of clinical trials at sites such as ClinicalTrials.gov. Such registration both improves transparency concerning what clinical trials have been and are being performed and empowers patients to find relevant clinical trials.
 OVERVIEW OF TRADITIONAL CLINICAL TRIAL DEVELOPMENT PHASES
OVERVIEW OF TRADITIONAL CLINICAL TRIAL DEVELOPMENT PHASES
Traditionally, a new anticancer agent is tested in a three-step process, starting with a small dose finding trial (phase I), followed by a pilot efficacy trial (phase II), and culminating with a large comparative randomized trial (phase III). This development paradigm was created and established in the era of cytotoxic chemotherapies and continues to be used today in the era of often less-toxic (and possibly not dose-dependent) targeted therapies, with some adaptations and innovations that we discuss later. Here we review the traditional paradigm without technical details, which can be found in many excellent sources for clinical trial design and conduct.2,5
Phase I
The main objective of the phase I trial is to determine the maximum tolerable dose of an agent to be subsequently used in testing for efficacy. Although initially developed in the setting of drug testing, this concept has been adapted to test radiation alone and combined drug/radiation regimens. The basic conceptual approach is that of sequential dose increases, or escalation, in small patient cohorts until the treatment-related adverse event rate reaches a predetermined level or unexpected toxicity is seen. This stepwise testing in phase I trials determines what is known as the maximum tolerated dose (MTD), which is putatively the most effective level at which to evaluate efficacy.
The key parameters to be defined at the outset of a phase I trial include patient eligibility criteria, starting dose and schedule of dose escalation (which should frame the expected MTD), events comprising the adverse event/toxicity response and the expected MTD, and finally the escalation design plan.10 By far, the dominant design has been the so-called 3+3 approach, where cohorts of three patients are exposed to a given dose, and based on the outcomes in that cohort, either de-escalation, escalation, or additional enrollment takes place. There are a large number of other designs, one of which may be particularly suited for radiation therapy trials, as we discuss shortly. It should be appreciated that the MTD is a relative concept that can change over time. For example, hematologic toxicity may be less dose limiting today than it was before the development of bone marrow stimulators such as erythropoietin and filgrastim.
Phase II
The primary purpose of the phase II trial is to determine the response rate of the treatment, seeking early evidence of clinical activity. An important secondary purpose is to gather more robust adverse event information at the established dose. The primary efficacy end point of the phase II trial has traditionally been tumor response; however, duration of response, progression-free survival (PFS), and site-specific activity such as locoregional control are all relevant and increasingly used. Measures of patient survival are usually secondary end points in phase II trials because of the limited sample size and follow-up duration of these trials. In general, phase II trials usually are not designed to provide definitive evidence that the test treatment is superior to current options. In fact, phase II trials have traditionally been single-arm studies comparing against a benchmark historical response rate. Reliance on this nonconcurrent external control rate can be problematic.11–13 Furthermore, patient selection factors can influence the results—for example, overall response rates in single-institution phase II trials can be significantly higher than in multicenter studies or subsequent phase III controlled trials.14 Randomized phase II trials have historically had a role in multiarm trials aimed at selecting the best treatment(s) to take forward for further testing.15 More recently, they have become favored as a means of providing more reliable pilot efficacy data.16 However, the preferred approach is changing as described later.
Phase III
Phase III trials are randomized comparisons between a new treatment regimen that has already shown promise in phase I/II trials and the current best standard of care (i.e., the control). Randomization offers a critical advantage over nonrandomized studies. Specifically, randomization balances the distribution of prognostic factors between treatment arms and ensures that treatment is assigned independent of these factors, thereby minimizing or eliminating these effects when comparing outcomes by treatment. When randomization is combined with treatment blinding of patients, researchers, or both, then even subjective outcomes can be assessed with minimal bias. Additionally, in multicenter studies, randomization can balance any systematic bias of the treating physicians or institutions.
Phase III trials can address one of several types of primary questions. For example, a study can be designed to determine if standard treatment is better than best supportive care. More often, phase III studies are designed to compare a new treatment with the current standard treatment. Phase III studies can also compare two or three different regimens with each other, as well as with standard treatment. Finally, a trial may be designed to demonstrate that a given treatment option is not worse than another by more than a tolerable margin. These “equivalence” trials, more accurately referred to as noninferiority trials, play an important role in development of less invasive or less burdensome treatment regimens.
The primary end point of a phase III trial is most typically overall survival (time to death from any cause); however, other important clinical end points such as disease-free survival (DFS) are increasingly justified. Because these trials aim to definitively demonstrate benefit with respect to these end points, the number of participants and follow-up period required for phase III trials is much longer than in phase II trials. Important secondary end points can include locoregional control and other site-specific failure end points, adverse event profiles, and quality of life measures.
Phase IV
Phase IV trials are also known as a postmarketing surveillance trials. These trials involve the safety surveillance of a drug after it receives regulatory approval for standard use. The safety surveillance is designed to detect any rare or long-term adverse effects over a much larger patient population and longer time period than was possible during the phase I–III clinical trials.
 UNIQUE FEATURE OF CLINICAL TRIALS IN RADIATION ONCOLOGY
UNIQUE FEATURE OF CLINICAL TRIALS IN RADIATION ONCOLOGY
Therapeutic clinical trials in radiation oncology typically involve the introduction of new technologies (e.g., the use of stereotactic body radiation for a new indication) or more frequently the novel combination of radiation therapy with a systemic agent. There are unique challenges—both biologic and clinical—that characterize clinical trials in radiation oncology compared to those not involving radiation.
Response rate is frequently used in early-phase medical oncology trials to indicate activity; however, considering radiation therapy itself is highly effective at shrinking tumors, this end point is not useful in radiation trials. A more appropriate “activity” end point for radiation trials may be PFS, although this itself is often difficult to objectively assess. Modern imaging end points (such as fluorodeoxyglucose [FDG] uptake) show promise as early readouts of activity but still require vigorous validation for individual disease sites. Furthermore, efficacy and toxicity end points in radiation trials depend on multiple biologic factors, including size of the target, proximity of tumor to sensitive normal tissues, accuracy of target volume definition, degree of patient immobilization, dose of radiation, and fractionation scheme. Consequently, quality-assurance measures are an essential feature of radiation trials, especially in the multi-institutional setting.17 Inadequate quality assurance and lack of consistency in radiation delivery have led to the conclusions obtained from large, expensive clinical trials being questioned.18–24 As a recent example, in RTOG 9704, which evaluated postoperative adjuvant chemoradiation treatment of pancreatic cancer, subtle protocol violations in target definition influenced both toxicity and survival.25
In medical oncology trials, adverse events typically occur during or within days of completing treatment. In contrast, toxicity following radiation therapy follows a biphasic course, early (within 3 months of starting treatment) and late (months to years later). Late toxicity is typically irreversible and hence important in determining the tolerability of an experimental treatment. Utilizing long-term toxicity as the primary end point in clinical trials is not practical; however, it nonetheless is imperative to collect and report robust information on long-term outcomes from radiation therapy trials. In fact, even in phase I radiation therapy trials, the follow-up period can be significantly longer than for those evaluating chemotherapy. A recent approach to dose escalation in phase I trials that considers late toxicities when deciding whether to advance to the next dosing level is discussed later.26
A further difference relates to the population studied. In medical oncology early-phase trials, participants have often received several lines of treatment and lack further therapeutic options. In contrast, patients in phase I radiation trials typically receive full-dose radiation treatment, and subjects may be treatment naïve. Furthermore, phase I trials in medical oncology are frequently “first in human” experience for the agent; toxicity is unpredictable and pharmacokinetic studies essential. Conversely, most multimodality phase I trials in radiation oncology involve systemic agents that have already been through extensive clinical testing; systemic toxicity is known and pharmacokinetic studies unnecessary. The purpose of the trial is to define the extent of local toxicity within the radiation field; consequently, radiation oncology phase I trials are organ specific. A recent study demonstrated that in reality, radiation phase I trials rarely utilize first in human agents, are associated with qualitatively predictable toxicity, and are comparatively safe.27 Examples of contemporary trial design in radiation oncology are provided in Table 14.2.
 STATISTICAL ISSUES IN CLINICAL TRIAL DESIGN
STATISTICAL ISSUES IN CLINICAL TRIAL DESIGN
Patient Population Definition and Stratification
A key issue in a clinical trial is a well-defined patient population to which the potential therapy applies. This is typically defined in terms of traditional disease characteristics reflecting putative prognosis, such as stage or its components. Increasingly, tumor pathology or marker features may be included. In any case, these must be unambiguously defined. Because factors defining eligibility are often critically related to prognosis, any randomized comparative study may use a stratified randomization approach to ensure equal representation of prognostic risk among treatment arms. Stratification factors need to be limited to a reasonable number because the total number of strata equals the product of the number of categories for each. For example, in a recently completed RTOG prostate cancer trial, stratification factors consisted of two levels of prostate-specific antigen (PSA) (<4 vs. 4 to 20), three cell differentiation categories (well, moderate, poor), and nodal status (N0 versus NX). The possible combinations of these variables create 2 × 3 × 2 = 12 strata within which treatments are to be balanced in allocation.
TABLE 14.2 EXAMPLES OF CLINICAL TRIAL DESIGNS FROM THE RADIATION THERAPY ONCOLOGY GROUPA

Randomization
Randomization is used differently in various phases of development but serves a similar purpose, which is to render treatment groups similar with respect to factors other than treatment that can influence outcomes. In phase I trials, there typically are not comparative groups, although there are situations where parallel cohorts of patients are being evaluated. Thus, randomization into cohorts ensures that these groups can be compared later for response biomarkers or other factors of interest. In phase II trials, randomization has been used in two similar but distinct ways. First, so-called selection designs have been used to help decide which of several potential candidate treatments to take forward to further definitive testing.15 In these trials, interest is not in statistically significant differences between treatments but rather is in the ability to nominally rank candidates in terms of best potential efficacy. It can be shown that this approach has high probability of identifying the most likely superior arm, although at the cost of false-positive findings, particularly if misused.28 A second and more recent role of randomization in phase II trials is to provide evidence, albeit at a less stringent criteria, that a test treatment is indeed promising.16 In phase III, randomization is critical for definitive unbiased evaluation.
With regard to implementation, randomization assignments can be simple or, more commonly, implemented using blocking or dynamic approaches with respect to balancing treatment arms by key factors, such as stratification variables mentioned earlier. Several proven methods are available.5
It is important to note that investigators must protect against practices that can erode or nullify the benefits of randomization. First, any breach of the random assignment process has an irreparable effect on the validity of the trial. Second, a large number (or differential number per treatment arm) of patient withdrawals can make the validity of the comparison suspect. Similarly, differential follow-up and consequently ascertainment of patient status between treatment arms can bias the treatment effect estimate. Third, bias in assessment of outcomes can have a major impact on the estimated treatment effect; thus, objective outcome measures and blinding of treatment assignment become important. Treatment assignment blinding is not feasible for radiotherapy and most chemotherapy regimens but can be used for many agents. In either case, and in particular for studies that cannot be blinded (among patients or caregivers), unambiguous, objectively defined end points are essential. In cases where determination of the end point involves possible observer subjectivity, such as when reading a diagnostic scan to determine disease progression, keeping assessors unaware of treatment assignment may be necessary.
End Points
In clinical trials, end points must be unambiguously defined, be assessable and reproducible, and reflect the action of the intervention. Typically, there is a single primary end point in a clinical trial; however, there may be numerous secondary end points.
Traditionally, in phase II cancer trials, treatment activity has been defined in terms of reduction in tumor burden. The most recent criteria for measuring activity are known as the Response Evaluation Criteria in Solid Tumors (RECIST).29 The criteria require the identification of target and nontarget lesions at baseline and their largest single dimensions. Categories of response are then defined—for example, complete response (CR), or disappearance of all target and nontarget lesions and no new lesions; partial response (PR), or 30% or greater decrease in the sum of the longest diameter of all target lesions, no progression of nontarget lesions, and no new lesions; and progressive disease (PD), or 20% or greater increase in target lesions, progression of nontarget lesions, or the occurrence of new lesions. A patient not satisfying either response or progression criteria is classified as having stable disease. Those achieving either a CR or PR are typically defined as objective responders, and the proportion of patients responding is then the primary end point of interest. Although widely used, there has long been concern that response defined this way is an inadequate substitute for more clinically relevant and objective end points such as survival time. In one study, fewer than 25% of agents that produced tumor response were eventually found to extend survival in comparative trials,30 whereas another suggested that tumor response is a reasonable surrogate for survival extension.31 Additional problems with the use of response rates in phase II trials include subjectivity and lack of reproducible assessments.32
As mentioned earlier, response is not as frequently used when radiation therapy is the test question, and in any case, other discrete binary end points can readily be used. For example, the proportion free from a given event (i.e., proportion alive, proportion recurrence-free, etc.) at a fixed time landmark such as 2 years is a common and straightforward end point.
A more informative end point that is used in many phase II and most phase III trials is the elapsed time from trial entry until occurrence of some event. The most straightforward of these is overall survival time, or time to death from any cause. This simple end point does not depend on adjudication of cause of death and its attendant complexities and naturally corrects for both favorable and unfavorable consequences of treatment. Although it can be verified or even ascertained from public records because of its simplicity, active follow-up per protocol remains of paramount importance. Cause-specific survival end points may also be considered; however, as mentioned, assigning cause of death is not simple, and one must account for “other cause” deaths and whether these have any relationship to treatment. In addition, whenever cause-specific deaths or other site-specific failure end points are used, methods for appropriately dealing with competing risks are required.33,34
Other commonly used time-to-event end points include DFS (time to recurrence or death from any cause) or PFS (time to disease progression, possibly determined via imaging or other assessments at regular intervals), although definitions of these are not standardized (e.g., see Hudis et al.35), and the specific failure events comprising given end points should be carefully specified. The main advantage of using DFS or PFS is the more rapid rate of events, leading to a smaller required sample size. In many cancer types, benefit with respect to these end points does not necessarily imply subsequent lengthened survival, although they may still represent clinical benefit for patients. For other disease settings (e.g., adjuvant therapy in colon cancer), DFS is a reliable and well-accepted primary end point that is strongly correlated with survival.36 This raises the topic of so-called surrogate end points, which are end points on which treatment benefits can be reliably measured. Various biomarkers and clinical end points have been studied and proposed as surrogate end points in clinical trials. Prentice37 specified criteria that a surrogate end point must fulfill if it is used to substitute for a clinical end point: the therapeutic intervention must exert benefit on both the surrogate and the clinical end point; the surrogate and clinical end point must be associated; and the effect of intervention on surrogate end point must mediate the clinical effect. An example of a widely studied surrogate marker is PSA, applied either as a static measure or as dynamic measures (PSA velocity; PSA doubling time; time to PSA nadir, particularly useful after radiation therapy of the intact prostate) to assess time to biochemical failure in patients with nonmetastatic adenocarcinoma of the prostate.38,39 This continues to be a developing area, and there remain many caveats and cautions regarding surrogate end points in clinical trials.40
Statistical Power and Sample Size
The overarching design consideration in clinical trials is to obtain sufficient information about an intervention so that a reliable decision can be made regarding its further development or use. In the classical (e.g., frequentist) statistical hypothesis testing paradigm, one sets up a null hypothesis of no treatment effect and an alternative hypothesis (which one hopes to validate) indicating a treatment effect. The type II or β error equals the probability that a statistical test fails to produce a decision in favor of a treatment effect when in fact the treatment is superior in the population. The complement of this probability (1 – β) is referred to as statistical power and equals the probability of correctly deciding in favor of a treatment benefit. Statistical power depends on the other principal parameters considered when planning the trial, specifically the probability of incorrectly finding in favor of a difference when none exists (type I or alpha error, usually set to 0.05 or 0.01 by convention), the a priori specification of a treatment effect that is considered both realistic and clinically material, and of course the sample size. It is imperative that trials be designed to achieve adequate statistical power; typically, 0.80 to 0.90 is desirable so as not to obtain equivocal findings concerning the potential worth of new treatments under consideration. Studies with low statistical power can cause delay or even abandonment of the development of promising treatments, as well as waste valuable resources, not least of which is the participation and goodwill of patients.41 In contrast, a “negative” trial that does not find the test treatment to be superior, if adequately powered, is informative in that resources can be directed into other more promising alternatives.
Thus, sample size to satisfy the power desired for the specified effect of interest is the key calculation in phase II and III clinical trials. (Phase I trials do not rely on hypothesis-driven sample size calculations, and the sample size derives from the specific design used.) The specific sample size calculation depends on the end point, and technical details will not be provided here; however, the two most common types of end points can be summarized as follows.
For a discrete binary end point in a phase II trial—for example, responded or did not respond—sample size calculations are straightforwardly performed using formulas for comparison of proportions. In a single-arm study, one aims to compare the observed response rate for the new agent to some historical response proportion, p0, or the response rate achievable with standard therapy in the target population. The main objective is to determine whether there is sufficient evidence to conclude that the response rate for the new regimen is greater than p0. We designate pA as a response rate which, if true, would be clinically material. We test the null hypothesis, H0 : p = p0, against the alternative hypothesis HA : p = pA. The values of p0 and pA and (and more importantly the difference), along with the sample size, will determine the power of the study. Note that to detect a small improvement (say, ≤10%) requires a large sample size. For example, to detect an improvement from a historical value of 20% to 30% with 85% power, more than 120 subjects are required. In addition, the value for both p0 and pA must be realistic; it is of little value to design and carry out a study to detect an effect size pA – p0 that is unlikely to be realized, simply because it is compatible with the number of patients that can be recruited.
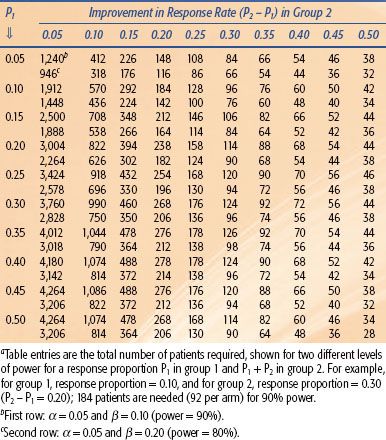
For two-arm randomized phase II trials with discrete end points, the previous discussion is simply redefined in terms of two-sample comparisons of proportions, and the sample size is consequently much larger. Table 14.3 shows some sample size requirements for various response differences, illustrating the influence of the effect size and power on the number required. Finally, if the end point is a fixed time landmark, such as proportion event free or alive at 1 year, then the estimates of the proportions may be derived from survival analysis methods to appropriately account for losses to follow-up.
In many randomized phase II and nearly all phase III trials, the time from randomization until occurrence of the event is of principal interest rather than the event status at some fixed time landmark. In larger phase II and phase III trials, recruitment may take place over a lengthy interval, with each patient having a different follow-up duration, and the use of follow-up time per patient is more efficient than waiting until all patients have reached some fixed time. The treatment effect measure is then specified in terms of failure hazards, which can be thought of as failure rates per unit of time. Hypotheses are thus usually formulated in terms of the hazard ratio (HR) as H0 : λA/λB = HR = 1.0, where λA and λB are the hazards for treatments A and B, versus the alternative, HA : HR <1.0, for some value of the HR that represents a clinically important difference in outcomes. Under the assumption that this ratio is relatively constant over time, a given HR can be converted to an absolute difference between groups in proportions remaining event free at a specific follow-up time. For example, a new/standard HR equal to 0.75, or a 25% reduction in failure rate in the experimental group relative to the standard group, may translate into an absolute difference in the proportion of patients remaining free from the event between groups of 4.6% at 5 years, if the standard group 5-year survival percentage is 80% (Table 14.4).
From the specification of difference of interest or effect size, then, the sample size in terms of number of events required to detect this difference with desired statistical power and significance level is determined. Depending on the anticipated accrual rate and the prognosis (e.g., rapidity of failure events) in the control treatment group, the number of patients required can then be approximated. The number of events required depends strongly on the HR, becoming dramatically larger as the HR approaches 1.0 (Table 14.4). The number of patients required and total duration of the trial depend on the rate of patient accrual and the failure rate in the control group, both of which contribute to the determination of how rapidly the requisite events will be observed. The accrual rate is typically estimated from previous experience and may also involve querying investigators to project the accrual rate per unit of time. Similarly, the failure rate for patients under standard therapy is derived from available data. The final computations are straightforward but generally require computer programs,42 although under certain assumptions can be approximated.43 Sample size methods have been extended to take into account other factors that will influence power, such as patients withdrawing from treatment (dropout), switching from the assigned treatment to the other group (crossover), or deviating from protocol treatment (noncompliance).44–46
TABLE 14.3 SAMPLE SIZE FOR A TWO-ARM COMPARATIVE (1:1 ALLOCATION) TRIAL WITH A BINARY END POINTA