- Type 1 diabetes is an immune-mediated disorder that leads to destruction of the islets of Langerhans causing profound insulin deficiency.
- Metabolic changes observed in type 1 diabetes are secondary to insulin deficiency interacting with environmental influences such as diet and exercise.
- The pathogenesis of type 2 diabetes mellitus is complex and incompletely understood. It appears to be caused by an interaction of genetic and environmental factors that lead to defects in insulin secretion, insulin action and glucose effectiveness.
Introduction
Hyperglycemia, for better or for worse [1], is the metabolic abnormality that has been used to define the presence of, and characterize, diabetes. Diabetes comprises a heterogeneous group of disorders characterized by fasting and/or post-prandial hyperglycemia. The underlying abnormalities that lead to the development of hyperglycemia, however, differ amongst subgroups. Conventionally, diabetes has been categorized into two subgroups that, from a metabolic standpoint, differ in the degree of insulin deficiency present. This broad dichotomy is simplistic as a given patient may exhibit metabolic abnormalities previously considered unique to each category [2].
Type 1 (T1DM), or immune-mediated diabetes, is characterized by evidence of immune-mediated destruction of the insulin-secreting β-cells in the islets of Langerhans. Usually this leads to absolute insulin deficiency, which is insufficient to prevent unrestrained lipolysis during systemic illness or severe physical stress. In type 2 diabetes (T2DM), however, although the endocrine pancreas can produce insulin, secretion and circulating concentrations of insulin are inappropriate for the prevailing glucose concentrations. T2DM has traditionally been considered a disorder of insulin signaling (exacerbated by poor diet, obesity and lack of physical activity) rather than a deficiency of insulin.
It is important to remember that obese patients with T1DM can behave in a fashion similar to patients with long-standing T2DM and that metabolic differences between the two categories may be more imagined than real.
Carbohydrate metabolism
In the fasting state, glucose appearance is determined by the rate of endogenous glucose release from the liver and to a lesser extent the kidney. This is collectively referred to as endogenous glucose production. Glucose concentrations increase when glucose appearance exceeds glucose disappearance and continues to increase until these rates are equal. In humans without diabetes, glucose concentrations average 4.5–5.5 mmol/L following a 6–12-hour overnight fast.
Gluconeogenesis is responsible for approximately 50–60% of endogenous glucose production following an overnight fast, with the proportion increasing with increasing duration of the fast [3]. Gluconeogenesis utilizes three-carbon precursors such as lactate, alanine and glycerol to synthesize glucose molecules.
Following an overnight fast, approximately 80% of glucose disposal is insulin independent and occurs in the brain, splanchnic tissues and erythrocytes [4]. The majority of insulin-mediated glucose disposal occurs in muscle [5]. Because insulin levels are low in the post-absorptive state, muscle predominantly uses free fatty acids (FFA) for fuel [6]. In the presence of low insulin concentrations, glucose taken up by tissues predominantly is oxidized or undergoes glycolysis to release alanine and lactate which can be re-utilized by the liver for gluconeogenesis [7].
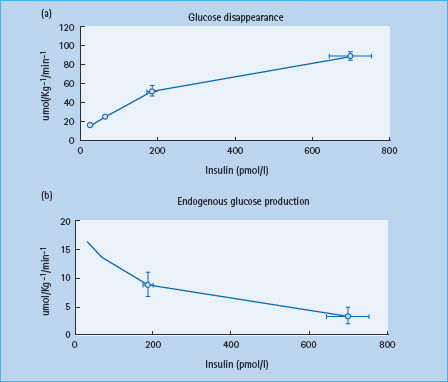
Sensitivity to insulin varies amongst tissues. Low concentrations of insulin limit lipolysis and prevent unrestrained breakdown of fat. The insulin concentrations sufficient to prevent lipolysis are insufficient to stimulate significant muscle glucose uptake. Whereas maximal suppression of endogenous glucose production occurs at insulin concentrations of approximately 250 pmol/L, these concentrations result in only half maximal stimulation of glucose uptake (Figure 13.1) [8].
Figure 13.1 Insulin dose-response curves for glucose production and utilization in non-diabetic subjects. Adapted from Vella A. Diabetologia 2002; 45 :1410-1415, with permission from Springer-Verlag.
Increases in plasma glucose, which occur within 5–10 minutes after eating stimulate insulin secretion and suppress glucagon secretion. The reciprocal changes in hepatic sinusoidal insulin and glucagon concentrations in concert with the elevated glucose concentrations enhance hepatic glucose uptake and suppress hepatic glucose production [9,10]. The splanchnic tissues initially extract 10–25% of ingested glucose and eventually dispose of approximately 40% of ingested glucose, with muscle accounting for most of the remainder [11]. These coordinated changes in hepatic and extrahepatic glucose metabolism generally limit the post-prandial rise in glucose to 7–8 mmol/L. Late post-prandial hypoglycemia is avoided by a smooth increase in hepatic glucose output to rates that closely approximate glucose uptake.
In the transition from normal glucose metabolism to overt diabetes, the relative contribution of alterations in glucose disappearance or appearance is uncertain. Most [12–15] but not all [16] epidemiologic studies that have attempted to elucidate the pathogenesis of impaired fasting glucose (IFG; defined as a fasting glucose of 5.8–6.9 mmol/L) have reported that insulin action is decreased in individuals with IFG. Weyer et al. [15] reported that fasting endogenous glucose production was increased in people with IFG. Bock et al. [17] subsequently established that insulin-induced suppression of endogenous glucose production and gluconeogenesis are impaired in people with IFG indicating hepatic insulin resistance. Epidemiologic studies have shown that 20–30% of people with IFG will develop frank diabetes within 5–10 years [18,19]. Indeed, subjects with a fasting glucose between 5.3 and 5.7 mmol/L have an 8% risk of developing diabetes within the next 10 years.
Regulation of glucose concentrations after meal ingestion is more complex. The pattern of change of post-prandial plasma glucose concentrations is determined by the extent to which glucose entering the systemic circulation (equal to the sum of endogenous glucose production and the systemic appearance of ingested glucose) exceeds or is exceeded by the rate at which glucose leaves the systemic circulation (glucose disappearance). Therefore, differences in post-prandial glucose concentrations could theoretically arise because of differences, alone or in combination, in rates of meal glucose appearance, suppression of endogenous glucose production or stimulation of glucose uptake [20,21].
Post-prandial hyperglycemia is primarily caused by reduced post-prandial glucose disappearance because suppression of endogenous glucose production and the rate of appearance of ingested glucose do not differ in people with IFG and normal fasting glucose [17]. The insulin secretion in response to higher post-prandial glucose concentrations is impaired in the subjects with IFG with accompanying defects in glucose disappearance and consequent post-prandial hyperglycemia. A further reduction in insulin secretion eventually results in overt T2DM [17].
Endogenous glucose production is regulated (inhibited) by insulin which increases hepatic glucose uptake by stimulating glucokinase activity and decreases hepatic glucose release by decreasing the conversion of glucose-6-phosphate to glucose. This latter step is regulated by glucose-6-phosphatase. Insulin can also stimulate glycogen synthesis, inhibit glycogen breakdown and suppress gluconeogenesis. Post-prandial hyperglycemia and hyperinsulinemia stimulate hepatic glycogen synthesis thereby replenishing hepatic glycogen stores. Hepatic glycogen synthesis occurs via both the direct (i.e. glycogen synthesis utilizing glucose-6-phosphate derived directly from extracellular glucose) or indirect pathway (i.e. glycogen synthesis utilizing glucose-6-phosphate derived from gluconeogenesis). The relative contribution of these two pathways appears to be determined by multiple factors including the duration of fast, composition of the meal and the prevailing insulin and glucagon concentrations [22,23].
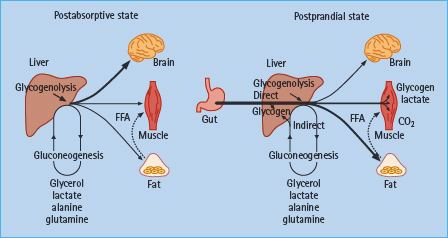
In the presence of euglycemia, rising hepatic sinusoidal concentrations of insulin suppress endogenous glucose production by decreasing glycogenolysis. Insulin concentrations within the physiologic range in healthy humans do not appreciably suppress gluconeogenesis and direct glucose-6-phosphate (derived from gluconeogenesis) into glycogen (Figure 13.2) [24].
Figure 13.2 Major sites of glucose metabolism in the post-absorptive and post-prandial states. After meal ingestion, the primary site of glucose uptake shifts from insulin-independent organs to insulin-dependent tissues. Gluconeogenic substrates are derived predominantly from peripheral tissues. Hepatic glycogen synthesis may occur via the direct or indirect (gluconeogenesis) pathways. Adapted from Dinneen et al. [21]. Copyright © 1992 Massachusetts Medical Society. All rights reserved.
Carbohydrate metabolism in type 1 diabetes
T1DM is characterized by insulin deficiency as a result of autoimmune destruction of the pancreatic β-cells. Fasting hyperglycemia does not develop until most (>80%) of β-cells are lost to the underlying autoimmune process. Defects in insulin secretion, however, are evident years before the development of diabetes in asymptomatic affected individuals. For example, siblings of people with T1DM who are islet antibody positive (a group at high risk for the development of T1DM) frequently exhibit a decreased first-phase of insulin secretion in response to intravenous glucose injection [25,26]. This usually occurs at a time when the response to other stimuli such as oral glucose or mixed meal ingestion is intact, suggesting that incretins and other secretagogues are capable of compensating for decreased islet cell mass.
Impaired insulin secretion is frequently accompanied by impaired insulin action [27,28]. The severity of insulin resistance is related to the degree of glycemic control. People with poorly controlled T1DM may exhibit the same degree of insulin resistance as people with T2DM [28,29]. In people with T1DM, the defect in insulin action is tissue-specific; for example, glucose uptake in cardiac muscle, as opposed to skeletal muscle, is normal [30]. Glucose oxidation and non-oxidative storage in people with T1DM is decreased in proportion to glucose uptake, suggesting that glucose transport and/or phosphorylation (after transport across the membrane) are the sites of defective insulin action [31]. In contrast, insulin-induced suppression of glucose production is not impaired and may in fact be increased in people with T1DM [32,33].
Insulin binding and action has been reported to be decreased in adipocytes [34,35] but normal in fibroblasts [36,37] of people with T1DM. This may be explained by the fact that insulin binding in adipocytes is measured immediately after biopsy while insulin binding to fibroblasts is measured following several days of culture. Decreased insulin binding in the former but not the latter suggests an effect of abnormal metabolic milieu rather than an intrinsic defect of insulin action. This is supported by several observations that improved chronic glycemic control is accompanied by improved whole-body insulin action [38,39]. It should be noted that when insulin action is measured by means of a hyperglycemic hyperinsulinemic clamp, hyperglycemia may compensate for small defects in insulin action by means of its ability (glucose) to stimulate its own uptake and suppress its own release (glucose effectiveness) [33].
Because insulin is typically delivered via the subcutaneous, rather than the intraportal route, treatment with insulin leads to systemic hyperinsulinemia which has been shown to impair insulin action in humans without diabetes [40]. The improved insulin action observed in people with T1DM following treatment with insulin, however, suggests that any negative effects of systemic hyperinsulinemia on insulin action are more than offset by the lowering of glucose concentrations and the reversal of glucose toxicity.
In the absence of insulin-stimulated muscle glucose uptake, substantial glucose disappearance in insulin-deficient people with T1DM occurs by glucose excretion in the urine and by glucose uptake via non-insulin-mediated pathways [41]. Food ingestion does not result in a rise in insulin or a reciprocal decrease in glucagon concentration [42]. Because of this, the increase in splanchnic glucose uptake and the decrease in endogenous glucose production are not appropriate for the prevailing glucose concentration. Post-prandial glycogen synthesis is markedly decreased, with most of the glycogen being synthesized by the indirect gluconeogenic pathway [43,44].
Consequently, because of abnormal hepatic glucose handling, excessive amounts of glucose reach the systemic circulation. The excessive rise in post-prandial glucose concentrations is compounded by the low insulin concentrations and defective insulin action present in people with poorly controlled T1DM [41,45]. In contrast, the ability of glucose to stimulate its own uptake and suppress its own release (glucose effectiveness) is normal in T1DM and most post-prandial glucose disposal occurs predominantly via non-insulin mediated pathways and by glucose excretion in the urine [46].
Post-prandial glucose metabolism in the splanchnic and extra-splanchnic tissues can be almost completely normalized by insulin administration which increases circulating insulin concentrations and prevents the excessive rise in counter-regulatory hormones that accompanies insulin deficiency. Insulin administration also restores post-prandial suppression of glucose production and stimulation of glucose uptake to rates similar to those observed in subjects without diabetes [41,47].
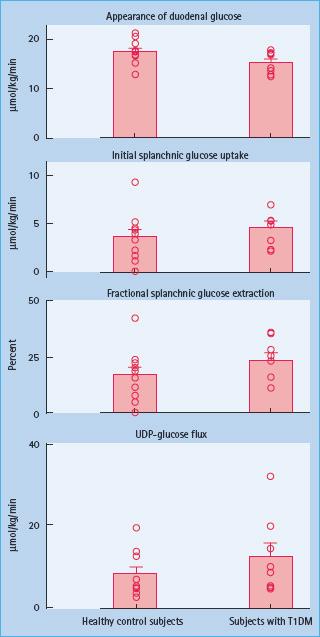
Animal studies have shown diabetes to be associated with hypertrophy of the intestinal mucosa and increased intestinal glucose transport [48,49]. By contrast, when glucose, insulin and glucagon concentrations are matched in individuals with T1DM and age- and weight-matched controls, initial splanchnic glucose extraction and uridine diphosphate (UDP)-glucose flux (an index of hepatic glycogen synthesis) do not differ between groups. This demonstrates that relative insulin deficiency, glucagon excess or both, rather than an intrinsic defect in splanchnic glucose metabolism are the primary causes of post-prandial hyperglycemia in people with poorly controlled T1DM (Figure 13.3) [33].
Figure 13.3 In the presence of matched glucose, insulin and glucagon concentrations initial splanchnic glucose extraction and UDP-glucose fl ux do not differ in individuals with type 1 diabetes and age-and weight-matched controls. FFA, free fatty acids; UDP-glucose, uridine diphosphate glucose. Adapted from Vella et al. [33].
Carbohydrate metabolism in type 2 diabetes
People with T2DM have elevated fasting glucose levels and excessive glycemic excursions following carbohydrate ingestion. Insulin secretion in those with T2DM is typically decreased and delayed following food ingestion [9,21]. Defects in insulin secretion are observed early in the evolution of T2DM. In fact, alterations in both the timing and amount of insulin secreted have been reported in relatives of patients with T2DM prior to the development of hyperglycemia [50,51].
Chronic hyperglycemia alone or in combination with elevated FFA impairs insulin secretion. Abnormalities in glucose sensing, insulin processing or intracellular signaling can alter insulin secretion [52]. In addition, β-cell mass decreases with increasing duration of diabetes [53,54]. Alterations in β- cell morphology occur in most people with T2DM with extensive intra-islet deposition of amylin commonly being observed [55,56].
Prolonged elevations in glucose concentration following ingestion of a carbohydrate-containing meal occur because post-prandial glucose appearance exceeds disappearance. The more significant the defects in insulin secretion and action, the higher glucose concentrations have to rise to balance glucose appearance and disappearance [57]. Glucose appearance is elevated because of failure to suppress hepatic glucose production because the systemic rate of appearance of ingested glucose does not differ from that observed in individuals without diabetes [9,11,17,58]. Although glucose disappearance is commonly higher in people with diabetes than in individuals without diabetes following a meal, this is in large part is accounted for by elevated rates of urinary glucose excretion. Furthermore, although elevated, the rates of glucose disappearance are not appropriate for the prevailing glucose concentrations [59,60].
Defects in insulin secretion and action both contribute to post-prandial hyperglycemia. A delay in the early rise in insulin concentrations causes a delay in suppression of glucose production, which in turn results in an excessive glycemic excursion. In contrast, a decrease in insulin action results in sustained hyperglycemia but has minimal effect of peak glucose concentrations. Whereas an isolated alteration in either hepatic or extrahepatic insulin action impairs glucose tolerance, a defect in both results in severe hyperglycemia [57].
Glucose is also an important regulator of its own metabolism. In the presence of basal insulin concentrations, an increase in plasma glucose stimulates glucose uptake and suppresses glucose production. The ability of glucose to regulate its own metabolism is impaired in T2DM. This is commonly referred to as a defect in “glucose effectiveness.” Whereas intravenous infusion of 35 g glucose in individuals without diabetes whose insulin concentrations are clamped at basal levels produces only a modest rise in plasma glucose concentration, infusion of the same amount of glucose results in severe hyperglycemia in people with T2DM [61]. The excessive rise in glucose is caused by impaired glucose induced stimulation of glucose uptake because glucose induced suppression of glucose production is normal [61,62].
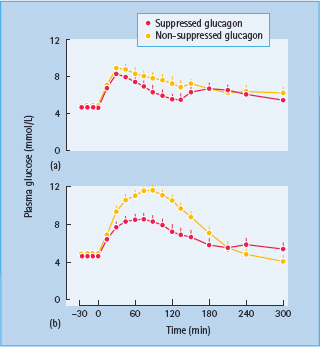
Inhibition of glucagon secretion lowers both fasting glucose and post-prandial glucose concentrations. Failure to suppress glucagon secretion appropriately, however, has minimal effect on glucose production and glucose tolerance when insulin secretion is intact [63,64]. In contrast, it causes marked hyperglycemia when insulin secretion is decreased and delayed, as is typical of T2DM. Taken together, these data suggest that agents that simultaneously improve insulin secretion, insulin action, glucose effectiveness as well glucagon secretion are likely to have a profound effect on glucose metabolism in people with T2DM (Figure 13.4) [64,65].
Figure 13.4 Healthy subjects received a non-diabetic (a) or a diabetic (b) insulin profi le on two occasions. On one occasion glucagon was infused in a manner that replicated the fall in glucagon concentrations that normally occurs after meal ingestion. On the other occasion glucagon was infused constantly to replicate the lack of glucagon suppression observed in type 2 diabetes in response to meal ingestion. Glycemic excursion was signifi cantly higher in the absence of glucagon suppression in subjects receiving an insulin profi le similar to that observed in patients with type 2 diabetes. Adapted from Shah et al. [64], with permission from the American Physiological Society.
Amylin is a 37 amino acid polypeptide that is co-secreted with insulin by the pancreatic β-cells in response to nutrient stimuli and other secretagogues. Human studies have shown that the plasma concentrations of amylin and insulin rise and fall in parallel in both the fasted and fed states [66]. Because amylin is potentially toxic to β-cells, it has been suggested that excessive amylin secretion may contribute to β-cell destruction in T2DM [67,68].
The secretion of incretins such as glucagon-like peptide 1 (GLP-1), in response to meal ingestion is decreased in T2DM [69–71]. Supraphysiologic concentrations of GLP-1, achieved by either intravenous infusion or subcutaneous injection, lower both fasting and post-prandial glucose concentrations in people with T2DM. GLP-1 does so by increasing insulin secretion, inhibiting glucagon secretion and delaying gastric emptying [72–75]. By contrast, GLP-1 does not appear to alter insulin action or glucose effectiveness in T2DM [76].
In addition to defects in insulin secretion, people with T2DM commonly exhibit defects in insulin action. Numerous studies have shown that insulin-snduced stimulation of glucose uptake in muscle and adipose tissues as well as insulin-induced suppression of glucose production are impaired in T2DM [77,78]. The severity of insulin resistance is influenced by multiple factors including exercise, obesity and diet as well as genetic factors. Insulin resistance increases with increasing severity of diabetes and improves but is not normalized by improved glycemic control [79]. Defects in the ability of insulin to regulate muscle and fat glucose metabolism are evident in normoglycemic relatives of people with T2DM strongly implying a genetic basis for at least some degree of insulin resistance [50,80].
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


