Introduction
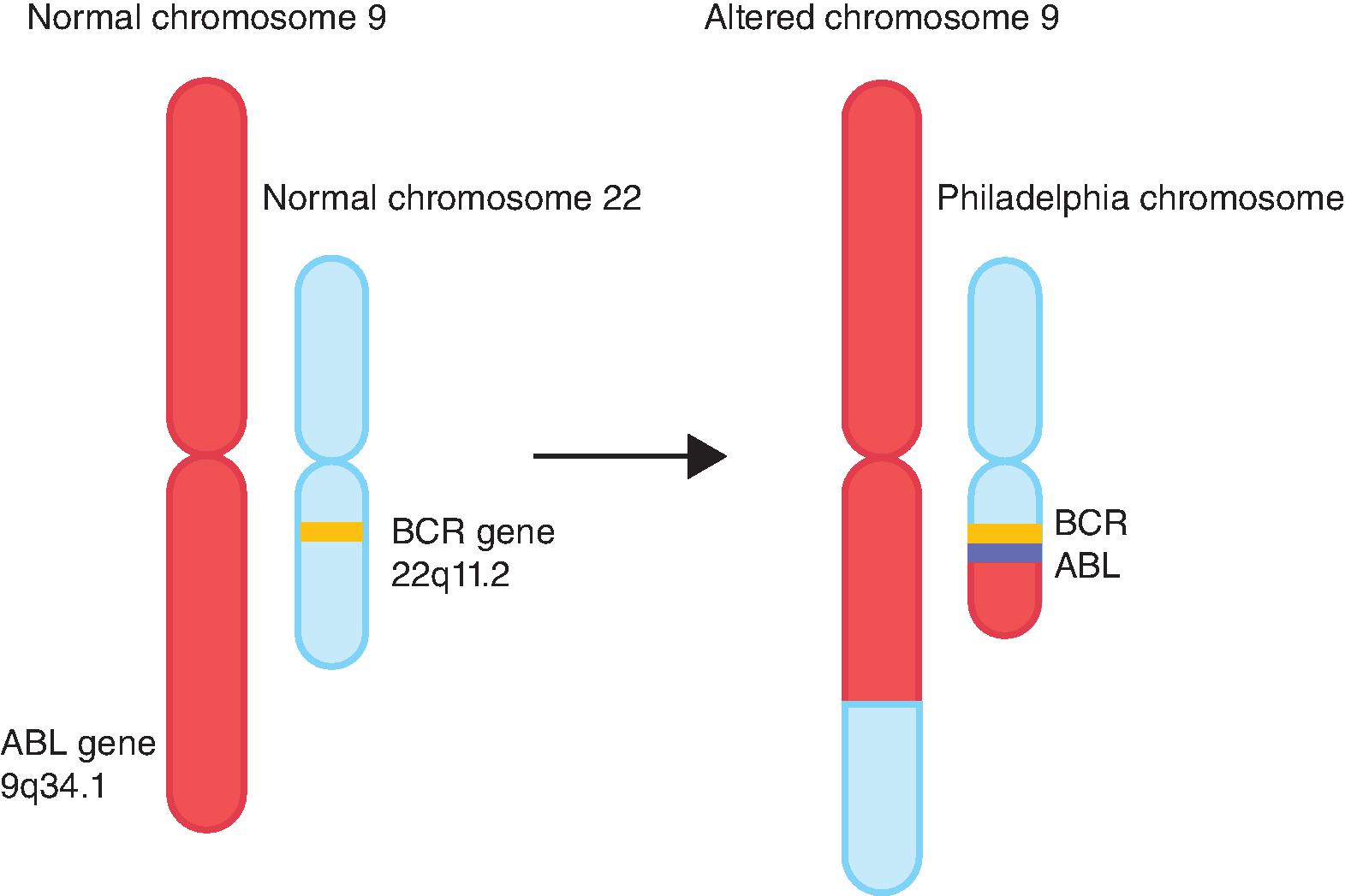
Over the last several years, molecularly targeted therapy has emerged as a new generation of cancer treatment and has been integrated into treatment protocols of many hematologic and solid tumors, either as monotherapy or in combination with chemotherapy. In general, targeted therapies include monoclonal antibodies and small molecule inhibitors that specifically affect the activity of genes or proteins mediating carcinogenesis. As described by Hanahan and Weinberg, cancer cells gain several hallmark features as they develop into invasive tumors. The acquisition of these features is achieved by deregulation of physiologic pathways for proliferation, growth, apoptosis, and angiogenesis. This is achieved mainly by genetic or transcriptional alterations, including chromosomal translocations, somatic mutations, gene amplification, overexpression, and/or downregulation of target proteins or enzymes. Understanding of the molecular events underlying the process of tumorigenesis, coupled with the advent of advanced testing options, has allowed the specific targeting of cancer cells harboring such alterations and the sparing of normal cells lacking them. This has translated into the development of several targeted therapy agents which have become integral parts of cancer treatment. Imatinib inhibits BCR-ABL translocation and is the prototype of successful application of targeted therapy in chronic myelogenous leukemia (CML) ( Fig. 12.1 ). This chapter will introduce molecular targets and their mechanisms of deregulation in cancer as they relate to the development of toxicities.
Receptor Tyrosine Kinases (RTKs)
- ■
Mechanism: RTKs are transmembrane proteins that play integral roles in the cellular signaling pathways involved in growth, proliferation, differentiation, metabolism, and motility. Deregulation of these receptors and/or their downstream effectors occurs in many cancers, making them attractive molecular targets in treatment. RTKs share a basic structure composed of an extracellular ligand-binding domain, a transmembrane α-helical domain, and a cytoplasmic domain. The latter includes a juxta-membrane region, a conserved tyrosine kinase domain, and carboxy-terminal region. The kinase domain catalyzes the phosphorylation of tyrosine residues on itself (autophosphorylation) and on other signaling molecules using adenosine triphosphate (ATP) as a substrate. First, extracellular ligands including growth factors, cytokines, and hormones bind specifically to their monomeric receptors, which promotes the formation of receptor dimers (homodimers or heterodimers). Unlike other RTKs, the insulin receptor (IR) preexists in a dimeric state, which is stabilized by ligand binding. The conformational change induced by ligand binding causes release of cis-autoinhibition and allows trans-autophosphorylation of tyrosine residues in the cytoplasmic domains. This induces receptor catalytic activity and generates docking sites for downstream cytoplasmic signaling proteins that possess Src homology-2 (SH2) or phosphotyrosine-binding (PTB) domains. This, in turn, leads to the activation of multiple signal transduction pathways, including the RAS/mitogen activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT), and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathways, which translates the extracellular signal into gene transcription and cellular response.
- ■
Alterations in malignancy: Aberrant activation of RTKs is a key event in the transformation of many tumors. The main mechanisms include gain-of-function mutations, chromosomal translocations resulting in oncogenic fusion genes, gene amplification, protein overexpression, and increased autocrine stimulation. RTKs most commonly implicated in cancers include EGFR (epidermal growth factor receptor), HER2, FGFR (fibroblast growth factor receptor), IGFR (insulin-like growth factor receptor), VEGFR (vascular endothelial growth factor receptor), PDGFR (platelet derived growth factor receptor), MET (hepatocyte growth factor receptor), ALK (anaplastic lymphoma kinase), KIT, and RET (rearranged during transfection).
- ■
RTK inhibitors: RTK inhibitors fall under 2 main categories: Monoclonal antibodies (mAbs) and small molecule tyrosine kinase inhibitors (TKIs). MAbs bind to the extracellular domain (ECD) of RTKs by competing with endogenous ligands, preventing ligand-induced receptor activation and inhibition of downstream signaling. This also leads to immune destruction of tumor cells by antibody-mediated and complement-mediated cytotoxicity, and antibody-mediated phagocytosis. TKIs are generally orally administered, low-molecular-weight molecules that cross the plasma membrane and act on the cytoplasmic side to inhibit tyrosine kinase activity, thus preventing autophosphorylation and activation of downstream signaling pathways. There are three types of TKIs; Type I TKIs compete with ATP for binding to the ATP binding site. This is the most common type. Many of these lack specificities and inhibit multiple RTKs owing to conservation of the ATP-binding site. Types II and III TKIs inhibit TKI activity by inducing conformational changes in RTKs independent of ATP competition. RTKs have been developed that have been successfully targeted in cancer therapy. Some of these targets include EGFR, HER2, VEGF/VEGFR, ALK, MET, RET, and KIT. Toxicities associated with these targets are largely due to inhibition of these targets in normal tissues.
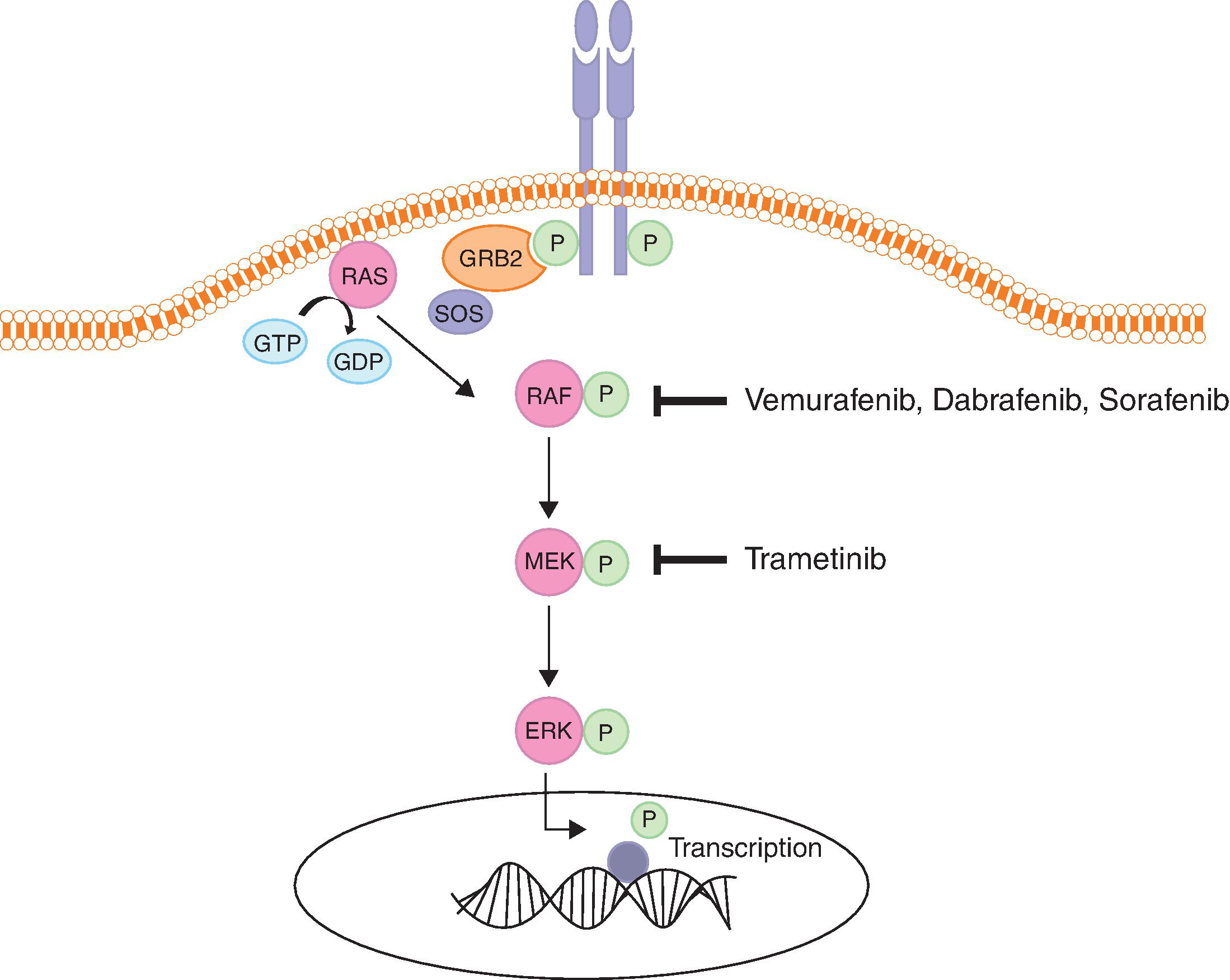
RAS/RAF/MEK/ERK Pathway
- ■
Mechanism: The RAS/RAF/MEK/ERK pathway is a key pathway involved in growth and proliferation and is deregulated in many cancers. RAS, RAF, MEK, and ERK are the main players in this pathway. RAS is a guanosine triphosphate (GTP)–binding protein with intrinsic GTPase activity that exists in two states, the inactive GDP-bound state (RAS-GDP), and the active GTP-bound state (RAS-GTP). Cycling between these two states is regulated by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs). GEFs, such as SOS (son of sevenless), promote GDP dissociation and GTP binding. GAPs, such as neurofibromin 1 (NF1) augment the GTPase activity of RAS, causing GTP hydrolysis and resulting in the inactive state. Signaling is initiated when growth factors or cytokines bind cell surface receptors such as RTKs or G-protein coupled receptors. This creates docking sites for adaptor proteins such as growth factor receptor-bound protein (GRB2) and SRC homology 2 domain-containing protein (SHC), which recruit GEFs like SOS to the cell membrane, resulting in RAS activation through exchange of GDP for GTP. This process stimulates dimerization and activation of RAF proteins, which are cytoplasmic serine/threonine kinases. RAF phosphorylates and activates MEK1 and MEK2 proteins, which are dual tyrosine and serine/threonine kinases. This in turn leads to phosphorylation and activation of ERK1 and ERK2 kinases, which phosphorylate cytoplasmic substrates and translocate to the nucleus to phosphorylate and activate transcription factors. The end result of this is activation of genes involved in survival, cell cycle progression, differentiation, apoptosis, migration, and feedback control of the pathway ( Fig. 12.2 ). ,