Mechanisms of Genomic Instability
David J. Gordon
David A. Barbie
Alan D. D’Andrea
David Pellman
Cancer arises from a series of genetic alterations that promote resistance to apoptosis, selfsufficiency in growth, cellular immortalization and escape from cell-cycle exit. The acquisition of these properties ultimately facilitates angiogenesis, invasion, and metastasis.1 It has been recognized for more than a century that genetic instability might represent an important pathway for the development of these disease characteristics. Von Hansemann2 identified abnormal mitotic figures in cancers, leading Boveri3 to propose that genetic instability, manifest in his experiments as whole-chromosome aneuploidy, could have a causal role in tumor development. The recognition that mutation of genes involved in monitoring genomic integrity underlies inherited cancer syndromes such as hereditary nonpolyposis colon cancer (HNPCC) and familial BRCA-mutant breast cancer provides clear evidence that genomic instability due to a so-called mutator phenotype can be the starting point for tumor development.4,5
However, many important questions remain. Does genomic instability play a central role in oncogenesis in common sporadic tumors? When during tumorigenesis does genetic instability develop, and what are the dominant mechanisms in specific cancer types? Why do inherited mutations in caretaker genes such as BRCA1 and BRCA2 lead to breast and ovarian cancer when their repair function is presumed to be ubiquitous? What is the relative contribution of telomere shortening to the development of genomic instability? Finally, what is the specific role of aneuploidy in cancer development, and what are the defects that promote chromosomal instability?
This chapter will outline the basic mechanisms involved in the maintenance of genomic integrity and will address these questions. One theme that has emerged from recent work in this area is that the development of genomic instability during cancer progression involves evolutionary tradeoffs.6,7,8 Loss of genetic stability is expected to increase the rate of growth-promoting or survivalpromoting mutations that could drive tumor growth. However, genomic instability will also increase the rate of deleterious mutations that could kill cells before they develop into tumors. Understanding how these factors balance out will ultimately be the key to understanding tumor development via genome destabilization. Perhaps most importantly, understanding this balance may also have implications for cancer therapeutics. If deleterious, genome-destabilizing mutations are found in the population of developing cancer cells, these defects may provide an “Achilles heel” for therapeutic attack.
BASIC DEFENSES AGAINST GENOMIC INSTABILITY
The roughly 1014 cells in the human body are continually exposed to sources of genomic injury, both spontaneous injury accompanying normal cell division and metabolism and external sources of damage. In addition to the oxidative stresses that are a by-product of cellular metabolism, cell populations that undergo constant turnover are subject to errors that may arise during the processes of DNA replication, mitosis, and telomere maintenance. Cells are also exposed to a variety of exogenous genotoxic insults. Examples include ultraviolet and gamma irradiation and certain chemicals (as detailed in other chapters). As a result, mechanisms have evolved at a number of different levels to guard against genomic instability and prevent the propagation of cancer-promoting and/or deleterious mutations. At an organismal level, tissues are designed to prevent the accumulation of cells with sustained disruption of genomic integrity.4 For example, those cell types in constant contact with the outside world, including the skin, gastrointestinal tract, and bronchial epithelium, undergo continuous self-renewal, with shedding of those differentiated cells that are exposed most directly to a potentially deleterious environment. In addition to being shielded from this stress, the stem cell compartment undergoes cell division fairly infrequently, with the bulk of exponential growth occurring in transit-amplifying cells that are ultimately discarded at the surface. Thus, in tissues such as the colon, stem cells are normally protected within the crypts, and those cells that proliferate and migrate toward the lumen are ultimately
eliminated. Nevertheless, this process is imperfect and subject to persistence of dysplastic clones if a cell sustains a mutation that affords a proliferative advantage.
eliminated. Nevertheless, this process is imperfect and subject to persistence of dysplastic clones if a cell sustains a mutation that affords a proliferative advantage.
Many cells also possess physiologic characteristics that can shield them from genotoxic injury.4,9 For example, melanin in the skin absorbs ultraviolet radiation, while antioxidants and enzymes such as catalase and superoxide dismutase reduce concentrations of reactive oxygen species generated as a result of cell metabolism. In addition, cytochrome P-450 enzymes detoxify a variety of chemicals, and glutathione-S-transferases (GSTs) conjugate glutathione with electrophilic compounds, neutralizing their mutagenic potential. Defective GST function has been observed in lung, breast, and prostate cancer and has been shown to predispose patients to myelodysplastic syndrome. Conversely, drugs that inhibit GST function are being tested in combination with chemotherapy in an attempt to enhance toxicity to cancer cells.
BARRIERS TO GENOMIC INSTABILITY
Cell Cycle Checkpoints
Coordinated progression through the cell cycle is crucial for the maintenance of genome stability.4,10,11 This is particularly the case for the main tasks of the cell cycle—DNA replication and mitosis. Either incomplete DNA replication or overreplication of DNA would generate lesions that could lead to chromosome breaks and rearrangements. Mitotic errors produce chromosome mis-segregation and whole-chromosome aneuploidy. These types of errors do not occur in isolation; a defect in one process can lead to a cascade of downstream events. Chromosome breaks can lead to translocations, chromosomes with two centromeres (dicentric chromosomes), anaphase bridges, and chromosome mis-segregation. Likewise, mitotic errors leading to aneuploidy will generate gene expression imbalances that could, in principle, compromise DNA replication, telomere maintenance, or DNA repair. Both DNA replication/repair and mitotic errors can cause cytokinesis to fail, resulting in tetraploid cells that contain extra centrosomes and are themselves genetically unstable. Although an extensive review of the cell cycle is beyond the scope of this chapter, selected features of the normal cell cycle that are crucial for preventing genome instability and cancer are described here. In particular, the following sections will focus on the restriction point, the DNA damage checkpoint, and the spindle assembly checkpoint. More extensive summaries of the eucaryotic cell cycle can be found in other chapters and in recent reviews.
Restriction Point
The decision to commit to cell division is controlled by a complex signaling system, the retinoblastoma protein (RB) pathway, that is the major target of human cancer-causing mutations.11,12 RB represses the transcription of genes involved in cell cycle progression by binding to the E2F family of transcription factors and altering the expression of E2F target genes, blocking E2F-mediated transactivation and recruiting active repressor complexes to promoters.13,14 E2F target genes include components of the nucleotide synthesis and DNA replication machinery that are essential for S phase entry and transit. In response to mitogenic signals during G1 phase of the cell cycle, RB is phosphorylated and inactivated by cyclin D/CDK (cyclin-dependent kinase) 4/6 complexes, followed by cyclin E/CDK2 and cyclinA/CDK2 complexes, resulting in E2F-target gene expression and S phase transit. Cyclin E/CDK2 regulates a number of other processes involved in the duplication of chromosomes, including the activation of histone gene transcription, as well as promoting the initiation of DNA replication and centrosome duplication.15,16 Deletion of cyclin E in mice results in defective endoreduplication, while constitutive overexpression of cyclin E has been linked to the generation of polyploidy and chromosomal instability. Thus, the RB pathway integrates intrinsic and external growth signals and is a key mediator of cyclin/CDK complexes that drive cell cycle progression.
CDK inhibitors and phosphatases provide other important mechanisms for counteracting the activity of CDKs and restricting cell cycle progression.4,11,16 CDK inhibitors fall into two general categories, including specific inhibitors of CDK4 such as p16INK4A, and those that target CDK activity more broadly such as p21 or p27. CDK2 is also targeted by inhibitory phosphorylation at its active site by the Wee1 family of protein kinases. Activation of cyclin E(A)/CDK2 during the normal G1/S phase transition thus requires the activity of the CDC25A phosphatase. Phosphorylation of CDC25A itself leads to its subsequent ubiquitinmediated proteasomal degradation and inhibition of S phase progression. Thus, expression of CDK inhibitors and down-regulation of CDC25A phosphatase activity are means by which checkpoint signals are able to mediate downstream cell cycle arrest. Furthermore, PP2A, a protein phosphatase that is also critical to the process of oncogenic transformation, has been shown to regulate an S phase checkpoint by dephosphorylating pRB and licensing recruitment of pRB to chromatin to suppress DNA replication.17
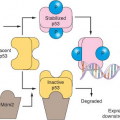
Heralded as the guardian of the genome, p53 integrates the response to DNA damage, replication stress, hypoxia, telomere dysfunction, and activated oncogenes and mediates downstream checkpoint activation.4,18,19,20 Inherited mutations in
p53 or its direct upstream activator CHK2 result in the Li Fraumeni cancer predisposition syndrome, and sporadic inactivation of p53 is one of the most frequent events observed in tumor development. Tumors lacking p53 exhibit widespread genomic instability resulting from an inability to arrest the cell cycle or trigger apoptosis in the setting of DNA damage and the cellular stresses previously described. In normal cells, p53 is maintained at low levels in the cytoplasm because of ubiquitination by MDM2 and proteasomal degradation. In response to checkpoint activation and phosphorylation, p53 increases in abundance and translocates to the nucleus, where it activates a transcriptional program that promotes cell cycle arrest, senescence, or apoptosis, depending on the cell type and conditions. The CDK inhibitor p21 is a key transcriptional target of p53 that mediates checkpoint arrest while repair is attempted. In response to a variety of signals p53 can trigger an apoptotic program, in part via transcriptional activation of proapoptotic targets such as NOXA and BAX.21
p53 or its direct upstream activator CHK2 result in the Li Fraumeni cancer predisposition syndrome, and sporadic inactivation of p53 is one of the most frequent events observed in tumor development. Tumors lacking p53 exhibit widespread genomic instability resulting from an inability to arrest the cell cycle or trigger apoptosis in the setting of DNA damage and the cellular stresses previously described. In normal cells, p53 is maintained at low levels in the cytoplasm because of ubiquitination by MDM2 and proteasomal degradation. In response to checkpoint activation and phosphorylation, p53 increases in abundance and translocates to the nucleus, where it activates a transcriptional program that promotes cell cycle arrest, senescence, or apoptosis, depending on the cell type and conditions. The CDK inhibitor p21 is a key transcriptional target of p53 that mediates checkpoint arrest while repair is attempted. In response to a variety of signals p53 can trigger an apoptotic program, in part via transcriptional activation of proapoptotic targets such as NOXA and BAX.21
DNA Damage Checkpoints
Activation of cell cycle checkpoints occurs as part of a larger DNA damage response pathway.22,23 There are three major DNA damage checkpoints, with two of the checkpoints occurring at the boundaries between G1/S and G2/M. The third checkpoint, however, occurs intra-S phase. In addition to promoting cell cycle arrest through the mechanisms described earlier, these pathways coordinate recruitment of repair proteins to the sites of DNA damage, modulation of transcription, activation of subsequent repair, and apoptosis. The signaling network that controls this response is initiated by the key DNA damage sensors, the ataxia-telangiectasia mutated (ATM) and AT and Rad3-related (ATR) protein kinases.10,11,24,25 ATM is principally activated in response to double-strand breaks, while ATR is activated by replication fork collapse and by bulky DNA lesions. As will be described in more detail later, both proteins phosphorylate multiple targets in coordinating the subsequent DNA damage response. Key signal transducers in this process include CHK2 (activated by ATM) and CHK1 (activated by ATR). p53 is a major substrate for ATM/CHK2 and ATR/CHK1 phosphorylation, and subsequent activation of p53 represents a principal mechanism by which cell cycle checkpoints are activated in response to DNA damage. Replication stress and hypoxia also appear to activate p53 through ATR signaling, while telomere dysfunction contributes to p53 activation through ATM (Fig. 2.1).18 In addition, a variety of stress responses have been shown to activate p38MAPK, which can promote checkpoint activation through both p53-dependent and -independent pathways.26,27 CHK2 activation also leads to phosphorylation and degradation of the phosphatase CDC25A, resulting in activation of an S phase checkpoint.28
Spindle Assembly Checkpoint
Errors that occur during mitosis are similarly monitored by a spindle checkpoint, which prevents progression into anaphase when chromosomes are improperly attached to the mitotic spindle.29 Key sensors of this response include the spindle checkpoint proteins, which assemble onto unattached kinetochores and generate a “wait anaphase” signal that prevents activation of anaphase effector proteins. This pathway is outlined in further detail later.
Cellular Senescence and Crisis
Cellular senescence is another mechanism that limits the progressive accumulation of cells with impaired genomic integrity and oncogenic potential.4,30,31,32 Originally described as an irreversible state of cell cycle exit in response to exhausted replicative potential of cultured cells, cellular senescence also occurs as a response to oncogene activation, oxidative stress, suboptimal culture conditions, and chemotherapy. The RB and p53 pathways have been shown to mediate the arrest by replicative senescence, whereby progressive telomere attrition elicits a DNA damage response similar to that induced by other genotoxic stresses (Fig. 2.1). In the setting of RB and p53 pathway inactivation, cells can bypass replicative senescence, but progressive telomere shortening results in the accumulation of massive genetic instability and a state of “crisis.” Most cells in crisis will die, but rare malignant clones can emerge. In humans, activation of the enzyme telomerase and subsequent maintenance of telomere length allows such cells to bypass crisis, resulting in cellular immortalization. By contrast, in mice, in which telomeres start out long and seldom shorten to a critical length, a similar but less well understood crisis event occurs that is, at least in part, related to differential sensitivity of mouse cells to oxidative damage in culture.
Senescence induced by oncogene activation, also known as oncogene-induced senescence, results from expression of p16INK4A and p14ARF (note that p14ARF in humans is p19ARF in the mouse), which exists in an alternative reading frame within the p16INK4A locus.33,34 p14ARF inhibits MDM2 function, in part by sequestering it in the nucleolus, resulting in the accumulation and activation of p53. p16INK4A expression is associated with both oncogene activation and genotoxic stress, promoting activation of RB and, in conjunction with HMGA chromatin proteins, the formation of stable heterochromatic foci that envelop and silence E2F target genes.35 Cellular senescence induced by this latter program is
refractory to RB and p53 inactivation, although it can be bypassed by inactivation of p16INK4A and HMGA proteins. Oncogene-induced senescence is also triggered by DNA replication stress, including prematurely terminated DNA replication forks, DNA double-strand breaks, and DNA hyperreplication.36,37 In a mouse model, inhibiting the DNA double-strand break response kinase ATM suppressed the induction of senescence and led to increased tumor size and invasiveness.
refractory to RB and p53 inactivation, although it can be bypassed by inactivation of p16INK4A and HMGA proteins. Oncogene-induced senescence is also triggered by DNA replication stress, including prematurely terminated DNA replication forks, DNA double-strand breaks, and DNA hyperreplication.36,37 In a mouse model, inhibiting the DNA double-strand break response kinase ATM suppressed the induction of senescence and led to increased tumor size and invasiveness.
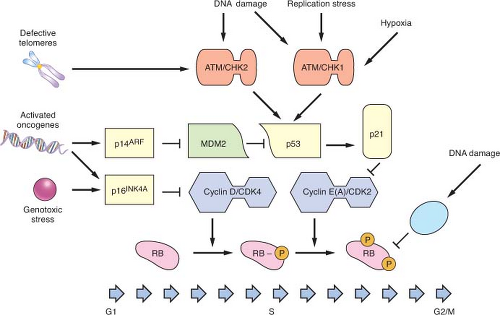 FIGURE 2.1 G1 pathways that can trigger cell cycle arrest, senescence, or apoptosis. A variety of threats to genomic integrity lead to activation of pathways that result in cell cycle arrest. Signaling via ATM/CHK2 and ATR/CHK1 leads to p53 activation, among other effects. One of the principle downstream effects of p53 is activation of p21 expression, with resultant cyclin E(A)/CDK2 inhibition and cell cycle arrest. Senescence, which results in a more sustained cell cycle exit, also involves the up-regulation of p14ARF and p16INK4A. Both proteins ultimately lead to retinoblastoma protein (RB) activation and G1 arrest via cyclin/CDK inhibition. In response to DNA damage during S phase, activation of PP2A can lead to dephosphorylation of RB and inhibition of DNA synthesis. |
Oncogene-induced senescence has been shown to occur in vivo, limiting tumor progression in models of lung and prostate cancer, melanoma, and lymphoma.33 Whereas inactivation of the PTEN tumor suppressor and resultant activation of the AKT signaling pathway in prostate epithelial cells appears to promote senescence through p14ARF, inappropriate activation of RAS signaling in other tissues results in p16INK4A-mediated senescence. Nonetheless, targeted activation of oncogenic K-RAS alleles in somatic tissues in mice predisposes to a wide variety of tumor types, including early-onset lung cancer, suggesting that this barrier may be readily overcome or that the consequence of RAS expression may vary depending on the context.38,39 Moreover, expression of endogenous levels of oncogenic K-RAS can promote proliferation, and it has been demonstrated that oncogene-induced senescence due to RAS activation can be dose-dependent.40,41,42
RAS-induced senescence in lymphocytes depends on heterochromatin formation via the Suv39h1 histone methyltransferase, the disruption of which facilitates lymphoma development in response to RAS activation.33 Furthermore, disruption of Suv39h1 by itself has been shown to disrupt heterochromatin formation and to promote genetic instability, contributing to lymphomagenesis.43 This may occur at least in part through cell division failure and the generation of unstable tetraploid cells (see later discussion). These findings confirm the in vitro observations that changes in chromatin structure contribute to cell cycle exit by senescence. In addition, they suggest that emerging epigenetic therapies such as histone deacetylase inhibitors and DNA methyltransferase inhibitors that interfere with chromatin silencing may disrupt this senescence barrier, a potential caveat to their use.
MUTATIONS IN CANCER
Despite multiple levels of protection against the development of genomic instability, with age, cells
can develop genetic alterations that escape detection.4 One path to genetic instability is inactivation of checkpoint proteins such as those previously described. The subsequent deregulation of the cell cycle and impairment of the response to genomic injury allows the progressive accumulation of lesions that can drive oncogenesis. Additionally, mutations can also occur in genes encoding the proteins that repair DNA damage and protect against the development of chromosome abnormalities. In this setting, accelerated mutation rates and chromosomal instability destabilize the genome and facilitate progression through the steps of oncogenic transformation. The basic types of genetic alterations observed in tumors, and the mechanisms by which genome destabilization can occur, are outlined in the next sections. Cancer predisposition syndromes that result from inherited defects in genome maintenance are highlighted in Table 2.1.
can develop genetic alterations that escape detection.4 One path to genetic instability is inactivation of checkpoint proteins such as those previously described. The subsequent deregulation of the cell cycle and impairment of the response to genomic injury allows the progressive accumulation of lesions that can drive oncogenesis. Additionally, mutations can also occur in genes encoding the proteins that repair DNA damage and protect against the development of chromosome abnormalities. In this setting, accelerated mutation rates and chromosomal instability destabilize the genome and facilitate progression through the steps of oncogenic transformation. The basic types of genetic alterations observed in tumors, and the mechanisms by which genome destabilization can occur, are outlined in the next sections. Cancer predisposition syndromes that result from inherited defects in genome maintenance are highlighted in Table 2.1.
TABLE 2.1 INHERITED GENOME MAINTENANCE DEFECTS WITH CANCER PREDISPOSITION | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
Point Mutations
Changes in the nucleotide sequence can arise from spontaneous mutation, exposure to endogenous or exogenous mutagens, or defects in the ability to detect and/or repair simple sequence errors.4,21 The spontaneous mutation rate per nucleotide per cell division has been estimated to be on the order of 10-9 in somatic cells and 10-11 in stem cells.44 Despite the remarkable fidelity of DNA polymerase and its inherent proofreading capacity, a variety of endogenous and exogenous chemical and radiation exposures can introduce additional DNA lesions, requiring the presence of multiple repair pathways for further protection of genomic integrity. Mutations arise when such errors are not detected and repaired by this machinery, which can occur when repair pathways are overwhelmed or defective. As a result, point mutations are frequently detected via sequencing of both oncogenes and tumor suppressor genes in multiple cancers. Notable examples include activating mutations in oncogenic kinases such as K-RAS in colorectal, pancreatic, and lung cancer, B-RAF in melanoma, and JAK2 in myeloproliferative disorders. In some instances, specific mutations have been linked to epidemiologic features such as tobacco exposure, with oncogenic K-RAS mutations in non-small cell lung cancer (NSCLC) occurring more frequently in smokers and epidermal growth factor receptor (EGFR) mutations in nonsmokers.45
Next-generation sequencing technology has ushered in a new era in cancer genomics.46,47 Massively parallel DNA sequencing platforms now allow for the routine sequencing of billions of bases of DNA per week and the identification of point mutations, insertions and deletions, copy
number changes, and genomic rearrangements on a genome-wide basis. Large-scale sequencing of coding sequences from a panel of colorectal and breast tumors, for example, revealed that these cancers harbor approximately 100 mutant genes, with computational methods predicting that 14 to 20 of these mutations will be bona fide tumor suppressor genes or oncogenes.48 In this unbiased effort, both known and unknown mutations were identified, with each tumor possessing a relatively unique cancer gene mutational signature. In another study, sequencing of coding regions of protein kinases in a large number of cancers identified “driver” mutations in approximately 120 genes across all samples.49 Although these studies identified a greater number of mutational events associated with oncogenesis than previously thought (the “state” of genome integrity), they do not necessarily imply a high “rate” of mutation, which remains low in most mature tumors.21
number changes, and genomic rearrangements on a genome-wide basis. Large-scale sequencing of coding sequences from a panel of colorectal and breast tumors, for example, revealed that these cancers harbor approximately 100 mutant genes, with computational methods predicting that 14 to 20 of these mutations will be bona fide tumor suppressor genes or oncogenes.48 In this unbiased effort, both known and unknown mutations were identified, with each tumor possessing a relatively unique cancer gene mutational signature. In another study, sequencing of coding regions of protein kinases in a large number of cancers identified “driver” mutations in approximately 120 genes across all samples.49 Although these studies identified a greater number of mutational events associated with oncogenesis than previously thought (the “state” of genome integrity), they do not necessarily imply a high “rate” of mutation, which remains low in most mature tumors.21
In two early studies, targeted gene resequencing was also applied to lung adenocarcinoma and glioblastoma multiforme (GBM) tumor samples.50,51 Both studies integrated the somatic mutation data with other genome-wide characterizations and clinical data. In the lung adenocarcinoma samples, for example, the sequencing data were used to identify multiple pathways, including MAPK signaling, p53 signaling, and the mTOR pathway, that are targeted by a combination of point mutations, copy number amplifications and deletions, and loss of heterozygosity (LOH). In a different study of GBM tumor samples, the sequencing of approximately 20,000 protein coding genes led to the discovery of a variety of genes that were not known to be altered in GBM, including the enzyme isocitrate dehydrogenase (IDH1).52 The cancer-associated IDH1 mutations result in the novel ability of the enzyme to catalyze the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG), which has been shown to lead to an elevated risk of malignant brain tumors.53 Subsequently, mutations in IDH1 have also been identified in acute myeloid leukemia (AML) genomes.54,55
More recently, there has been a rapid progression from targeted gene sequencing to targeted whole-genome and whole-transcriptome sequencing. The first sequencing of a whole cancer genome was reported for AML.56 Acquired mutations in coding sequences of annotated genes were identified in ten genes in the AML genome by comparing the genomic DNA of leukemia cells with normal skin cells obtained from a patient with FAB M1 AML. Two of the identified mutations were in genes previously described to have a role in leukemogenesis, FLT3 and NPM1. The other eight mutations, however, were in genes that were not previously implicated in the pathogenesis of AML. Four of the affected genes (PTPRT, CDH24, PCLKC, and SLC15A1), though, are in gene families that are strongly implicated in cancer pathogenesis. Intriguingly, the remaining genes (KNDC1, GPR123, EBI2, and GRINL1B) are involved in metabolic pathways.
The genomes of a small cell lung cancer, melanoma, and breast tumor have also been described.57,58,59 Interestingly, the mutations in both the lung cancer and melanoma genomes were not distributed evenly throughout the genome—many were present outside the gene-coding regions, suggesting that cells had repaired damaged DNA in those key regions. Sequencing of the melanoma genome, for example, revealed multiple levels of selective DNA repair, including the preferential targeting of repair to transcribed regions compared with nontranscribed regions, to exons compared with introns, to transcribed DNA strands compared with nontranscribed strands, and to the 5′ end of genes compared with the 3′ end.
Next-generation sequencing has also been applied to RNA (“RNA-Seq”) extracted from tumor cells for complete transcriptome characterization.60,61 This approach is more sensitive than microarrays and also provides data that can be used to evaluate for allele-specific expression, structural and copy-number alterations, alternative splice isoforms, fusion transcripts, and single nucleotide mutations.60 RNA-Seq, for example, was applied to four granulosa-cell tumors and identified missense point mutations in the FOXL2 gene, which encodes a transcription factor known to be crucial in granulosa cell development.62 In a different study, RNA-Seq was used to identify both known and novel fusion transcripts in prostate cancer samples.63 RNA-Seq has also been used to study the role of microRNAs in the regulation of gene expression in both normal and cancerous cells.
The number of sequenced cancer genomes is likely to expand substantially in coming years. The Cancer Genome Atlas Program of the U.S. National Cancer Institute, for example, initially focused its large-scale genomic analysis on only three tumor types, GBM, ovarian serous cystadenocarcinoma, and lung squamous carcinoma. The scope of the Cancer Genome Atlas Research Network, however, has now expanded to include more than 20 tumor types and thousands of samples. The clinical and translational implications of routine cancer genome sequencing are profound and include the identification of new drug targets, as well as the generation of new insights into the genetic patterns of disease phenotype, prognosis, and therapeutic response. One shortcoming of direct sequencing, though, is its failure to detect epigenetic changes, such as DNA methylation, which may alter gene expression indirectly.
Although the role of 2HG in cancer development remains unclear, the identification of this unexpected class of mutations validates the highthroughput cancer genome sequencing approach. Also, because the mutations in IDH1 result in a gain of function, there is much excitement about
developing small molecule inhibitors of mutant IDH1. Although major clinical impact of largescale sequencing projects is yet to be realized, the discovery of IDH1 illustrates the potential of this approach.
developing small molecule inhibitors of mutant IDH1. Although major clinical impact of largescale sequencing projects is yet to be realized, the discovery of IDH1 illustrates the potential of this approach.
 FIGURE 2.2 Common cytogenetic abnormalities. Metaphase spread derived from a mouse tumor model (combined telomerase and p53 deficiency). Individual chromosomes are highlighted by spectral karyotyping (SKY) using fluorescent chromosome probes (right panel). Normal mouse cells contain a diploid complement of 40 chromosomes. In this sample, more than 260 chromosomes are observed, with multiple dicentric chromosomes and nonreciprocal translocations. |
Translocations
Unlike point mutations, small insertions, or deletions of nucleotides, larger chromosomal changes such as translocations, amplifications, and deletions may be observed using cytogenetic analysis4,21 (Fig. 2.2). Chromosome translocation involves juxtaposition of two different chromosome segments, resulting in fusion of two different genes or placement of a gene next to an inappropriate regulatory element. Examples include t(9;22) in chronic myelogenous leukemia, resulting in expression of the growth promoting BCR-ABL gene product, and t(14;18) in follicular lymphoma, resulting in overexpression of the antiapoptotic protein BCL2 as a result of its fusion with the immunoglobulin heavy chain promoter. Recent work has also identified a translocation between the immunoglobulin heavy chain and the cytokine receptor CRLF2 in a subset of precursor B-cell acute lymphoblastic leukemia associated with a poor outcome and activating JAK mutations.64,65
One exciting recent development is that translocations not only create chimeric genes or alter promoter sequences, but can also affect the expression of microRNAs.66,67 MicroRNAs are short regulatory RNAs that control mRNA stability and/or translation, and changes in microRNA expression have been linked to prognostic factors and progression in diseases such as chronic lymphocytic leukemia.68
Although translocations and gene fusions are a hallmark of cancer, the mechanisms underlying their genesis are unclear. Recent work, however, has begun to elucidate the mechanisms of some tissuespecific translocations, such as TMPRSS2 to ERG and ETV1 in prostate cancer.69,70 A clever bioinformatics approach, termed cancer outlier profile analysis, was used to first identify this recurrent translocation in prostate cancer.71 This approach was used to successfully identify recurrent gene fusions of TMPRSS2 to ERG or ETV1 in prostate cancer. Further mechanistic work has shown that this translocation requires two roles of the androgen receptor.69 First, ligand-dependent binding of the androgen receptor to intronic binding sites near the tumor translocation sites creates specific intra- and interchromosomal interactions that result in the spatial proximity of tumor translocation partners. Second, the intron-bound androgen receptor alters local chromatin architecture and recruits the ligand and genotoxic stress-induced enzymes, including the activation-induced cytidine deaminase and LINE-1 repeat-encoded ORF2 endonuclease, to these regions, which results in the generation of DNA double-stranded breaks (DSB). The DSB are subsequently ligated by the nonhomologous end-joining (NHEJ) machinery to create translocations. Further elucidation of the mechanisms leading to gene- and tissue-specific translocations will advance the understanding of basic mechanisms of cancer, as well as possibly facilitating the development of new therapeutic strategies.
Amplifications and Deletions
Amplifications can be detected cytogenetically as double-minute chromosomes or regions of excess signal intensity using fluorescence in situ hybridization. Such “amplicons” may range in size from 0.5 to 10 megabases of DNA, resulting in multiple copies of both oncogenes and their neighboring
sequences. Conversely, deletions result in loss of chromosomal regions, and can involve small interstitial segments or entire chromosome arms. Genetic alteration of tumor suppressor genes frequently involves mutation in one allele and deletion of the second allele as part of a larger chromosomal segment, resulting in regions of uniform sequence with LOH.
sequences. Conversely, deletions result in loss of chromosomal regions, and can involve small interstitial segments or entire chromosome arms. Genetic alteration of tumor suppressor genes frequently involves mutation in one allele and deletion of the second allele as part of a larger chromosomal segment, resulting in regions of uniform sequence with LOH.
A recent study reported the high-resolution analysis of somatic copy-number alterations (SCNAs) from 3,131 cancer specimens belonging to 26 histologic types.72 The most prevalent SCNAs were either very short (focal) or almost the length of a chromosome arm or whole chromosome (arm level). The focal SCNAs occurred at a frequency inversely related to their lengths, with a median length of 1.8 megabases. Arm-level SCNAs occurred approximately 30 times more frequently than expected by the inverse-length distribution associated with focal SCNAs. This observation was seen across all cancer types and applied to both copy gains and losses. The study also identified 158 regions of focal SCNAs that were altered at significant frequency across several cancer types, of which 122 could not be explained by the presence of an oncogene located within the region. Several gene families were enriched among the regions of focal SCNA, including the BCL2 family of apoptosis regulators and the NF-kB pathway. Interestingly, the finding that most of the SCNAs were found in multiple cancer types suggests that the diversity across cancer genomes may reflect the combinations of a limited number of functionally relevant events.
Whole-Chromosome Loss/Gain
Nearly all solid tumor types exhibit whole-chromosome loss or gain, resulting in alterations in chromosome number or aneuploidy.21,73 As will be described later, such defects are generally the result of chromosomal mis-segregation during mitosis. Glioblastomas, for example, frequently exhibit loss of chromosome 10, inactivating the tumor suppressor PTEN, while melanomas often show gain of chromosome 7, from which B-RAF is expressed. Monosomy 7 and trisomy 8 are associated with myelodysplasia and AML. Whole-chromosome loss may be underestimated by karyotypic analysis, as loss of one parental chromosome may be accompanied by duplication of the other parental chromosome, resulting in an abnormal “allelotype” with accompanying LOH, or copy neutral loss of heterozygosity (CN-LOH).74
CN-LOH, also referred to as uniparental disomy, is common in cancer and has been described in AML, breast cancer, multiple myeloma, basal cell carcinoma, childhood acute lymphoblastic leukemia, chronic lymphocytic lymphoma, myelodysplastic syndrome, and glioblastoma.75 Furthermore, CN-LOH has been shown to have prognostic significance in a number of these cancer types, including primary and secondary AML.76 The specific role of CN-LOH in tumorigenesis remains undefined, but possible mechanisms include the duplication of oncogenes, loss of tumor suppressors, or acquisition of improper epigenetic patterns.77 In mice, it has been demonstrated that Bub1 insufficiency can drive tumor formation through tumor suppressor gene LOH.78 Specifically, Bub1 insufficiency predisposed p53+/− mice to thymic lymphomas and ApcMin/+ mice to colonic tumors. These tumors demonstrated CN-LOH and lacked the nonmutated tumor suppressor allele, but had gained a copy of the mutant allele.
Epigenetics
Significant evidence now indicates that epigenetic modifications, or heritable changes in gene expression that are not caused by changes in DNA sequence, are critical factors in the pathogenesis of cancer.79 Although beyond the scope of this chapter, epigenetic mechanisms controlling the transcription of genes involved in cell differentiation, proliferation, and survival are often targets for deregulation in the development of cancer. These epigenetic alterations include DNA methylation, covalent modifications of histones, and noncovalent changes in nucleosome position. The role of epigenetic modifications in cancer pathogenesis is illustrated by the tumor suppressor SNF5, which regulates the epigenome as a member of the SWI/SNF chromatin remodeling complex.80 Biallelic inactivation of SNF5 is found in the majority of malignant rhabdoid tumors. Most human SNF5-deficient cancers are diploid, lack genomic amplifications/deletions, and are genomically stable.81 Furthermore, the epigenetically based changes in transcription that occur following loss of SNF5 correlate with the tumor phenotype.
Recent work with an NSCLC cell line, PC9, has also identified a novel role for epigenetics in acquired drug resistance.82 Treatment of PC9 cells, which have an activating mutation in the EGFR, with the drug erlotinib results in the death of nearly all of the parental cells. A small percentage of the PC9 cells, however, demonstrate significantly reduced drug sensitivity and remain viable through activation of IGF-1 receptor signaling and an altered chromatin state that requires the histone demethylase RBP2. This drug-tolerant phenotype is transiently acquired at low frequency by individual cells within the population. The drug-tolerant subpopulation can be selectively ablated by treatment with IGF-1 receptor inhibitors or chromatin-modifying agents, such as HDAC inhibitors. There has been much discussion and controversy about the existence of drug-resistant cancer stem cells; the epigenetic effects described in this work provide a potential mechanism for the generation of some “cancer stem
cells.” Furthermore, this research suggests that the potentially reversible nature of epigenetic changes, unlike genetic mutations, may provide a unique therapeutic avenue in the treatment of cancer.
cells.” Furthermore, this research suggests that the potentially reversible nature of epigenetic changes, unlike genetic mutations, may provide a unique therapeutic avenue in the treatment of cancer.
MECHANISMS OF GENOME DESTABILIZATION IN HUMAN TUMORS
Microsatellite Instability
One of the earliest insights into the contribution of genome destabilization to carcinogenesis came from the study of the familial cancer syndrome HNPCC.4,11,83 It had been recognized that a subset of sporadic colon cancers and a majority of cancers derived from patients with HNPCC exhibited frequent mutations, particularly in regions of simple repeat sequences known as microsatellites. This type of genetic instability, termed microsatellite instability (MIN), had been described in bacteria and yeast mutants defective in mismatch repair. Linkage analysis in kindreds with HNPCC revealed germ line mutations in hMSH2 and hMLH1, which are human homologues of the mutL and mutS mismatch repair genes in Escherichia coli. It is now known that mutations in other components of the human mismatch repair process, hPMS2, and hMSH6, are also observed in families with HNPCC.
Mismatch repair corrects mispaired bases that can result from errors during DNA replication, as well as mismatched bases occurring in recombination intermediates or occurring as a result of some types of chemical damage to DNA.84 Mismatched bases are recognized by a complex of MSH2 and MSH6, recruiting MLH1 and PMS2 to the site to initiate the subsequent steps of repair, including excision, DNA synthesis, and ligation (Fig. 2.3). Larger insertion/deletion mispairs due to slippage of the replication machinery in repetitive sequences or recombination errors form a loop structure that is alternatively recognized by a complex of MSH2 and MSH3, with recruitment of an MLH1/MLH3 complex promoting subsequent repair. Cancer cells that exhibit MIN from defects in these components have a nucleotide mutation rate that has been estimated at two to three orders greater than that of normal cells. MLH1 and MSH2 have also been shown to have functions outside mismatch repair, as defects in these proteins have been associated with an impaired G2/M cell cycle checkpoint in response to alkylating agents as well as abnormalities in meiotic recombination in mouse knockout models.9
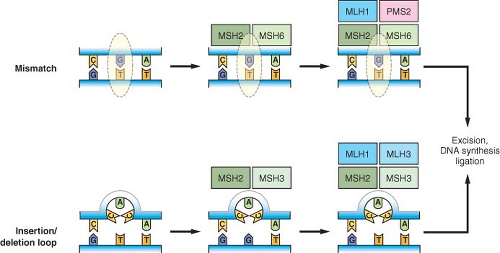 FIGURE 2.3 Mismatch repair pathways. Mispaired bases due to errors in DNA replication or other causes are recognized by the mismatch repair machinery. The initial step involves recognition of simple mismatches by MSH2 and MSH6 (upper panel), or recognition of insertion/deletion loops by MSH2 and MSH3 (lower panel). Subsequent steps involve recruitment of MLH1 and PMS2 to mismatch sites, or MLH1 and MLH3 to insertion/deletion loop sites. This is followed by excision of the respective lesions, DNA synthesis, and ligation to complete the repair. |
HNPCC is associated with a 60% to 80% lifetime risk of developing colorectal cancer and is responsible for 2% to 5% of all cases of colorectal cancer.85 Nearly 85% of HNPCC patients have mutations in MLH1 or MSH2, with median age at colon cancer diagnosis being significantly lower in MLH1 mutation carriers and in males, and with more frequent extracolonic tumors observed in MSH2 carriers. The MIN phenotype is also observed in 15% of sporadic colon cancers, often due to epigenetic silencing of mismatch
repair genes such as MLH1. Colorectal cancers exhibiting MIN are typically diploid, in contrast to the remaining 85% of cases, which are associated with chromosomal instability (CIN).86 Experimental evidence supports the idea that MIN occurs very early in sporadic colorectal cancer formation, prior to anaphase-promoting complex (APC) inactivation, increasing genomic instability and thus obviating the selection pressure to develop another mechanism of genomic instability, CIN. In both sporadic cases and HNPCC, MIN has been associated with more favorable prognosis, lack of p53 mutation, and a potential resistance to 5-fluorouracil chemotherapy. These observations are in accordance with the hypothesis that the accelerated mutation rate facilitates cancer evolution, but at the same time compromises fitness of cells, presumably by the accumulation of deleterious mutations.
repair genes such as MLH1. Colorectal cancers exhibiting MIN are typically diploid, in contrast to the remaining 85% of cases, which are associated with chromosomal instability (CIN).86 Experimental evidence supports the idea that MIN occurs very early in sporadic colorectal cancer formation, prior to anaphase-promoting complex (APC) inactivation, increasing genomic instability and thus obviating the selection pressure to develop another mechanism of genomic instability, CIN. In both sporadic cases and HNPCC, MIN has been associated with more favorable prognosis, lack of p53 mutation, and a potential resistance to 5-fluorouracil chemotherapy. These observations are in accordance with the hypothesis that the accelerated mutation rate facilitates cancer evolution, but at the same time compromises fitness of cells, presumably by the accumulation of deleterious mutations.
Nucleotide Excision Repair/Base Excision Repair Defects
Whereas the mismatch repair pathway functions primarily in the recognition of and repair of replication errors, the nucleotide excision repair (NER) and base excision repair (BER) pathways respond principally to lesions created by exogenous or endogenous DNA damaging agents.4




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
