Invasion and motility. Normal tissue requires proper adhesions with basement membrane and/or neighboring cells to signal to each other that proper tissue compartment size and homeostasis is being maintained. Tumor cells display diminished cellular adhesion, allowing them to become motile, a fundamental property of metastatic cells. Tumor cells use their migratory and invasive properties in order to burrow through surrounding extracellular stroma and to gain entry into blood vessels and lymphatics.
Intravasation and survival in the circulation. Once tumor cells enter the circulation, or intravasate, they must be able to withstand the physical shear forces and the hostility of sentinel immune cells. Solid tumors are not accustomed to surviving as single cells without attachments
and often interact with each other or blood elements to form intravascular tumor emboli.
TABLE 10.1 STEREOTYPIC PATTERNS OF METASTASIS TO DISTANT ORGANS BY CANCER TYPE
Cancer Type
Site of Metastasis
Breast carcinomas
Primarily bone, lung, pleura and liver; less frequently brain and adrenal. ER-positive tumors preferentially spread to bone; ER-negative tumors metastasize more aggressively to visceral organs.
Lung cancers
The two most common types of lung cancer have different etiologies. SCLC disseminates rapidly to many organs including the liver, brain, adrenals, pancreas, contralateral lung, and bone. NSCLC often spreads to the contralateral lung and the brain, and also to adrenal glands, liver, and bones.
Prostate carcinoma
Almost exclusively to bone; forms osteoblastic lesions filling the marrow cavity with mineralized osseous matrix, unlike the osteolytic metastasis caused by breast cancer.
Pancreatic cancer
Aggressive spread to the liver, lungs, and surrounding viscera.
Colon cancer
The portal circulation pattern favors dissemination to the liver and peritoneal cavity, but metastasis also occurs in the lungs.
Ovarian carcinoma
Local spread in the peritoneal cavity.
Sarcomas
Various types of sarcoma; mesenchymal origin; mainly metastasize to the lungs.
Myeloma
Hematologic malignancy of the bone marrow that causes osteolytic bone lesions, sometimes spreading to other organs.
Glioma
These brain tumors display little propensity for distance organ metastasis, despite aggressively invading the central nervous system.
Neuroblastoma
Pediatric tumors arising from nervous tissue of the adrenal gland. Forms bone, liver, and lung metastases, which spontaneously regress in some cases.
ER, estrogen receptor; SCLC, small cell lung cancer; NSCLC, non-small cell lung carcinoma.
Arrest and extravasation. Once arrested in the capillary system of distant organs, tumor cells must extravasate, or exit the circulation, into foreign parenchyma.
Growth in distant organs. Successful adaptation to the new microenvironment results in sustained growth. Of all the steps in the metastatic cascade, the ability to grow in distant organs has the greatest clinical impact and lies at the core of the seed and soil hypothesis. Accomplishing this step may be rate-limiting and may determine whether distant relapse occurs rapidly or dormancy ensues.
by mathematical considerations predicting that genetic changes accumulate faster with increased population size. Larger tumors are more likely to contain rare cells that are metastatically competent, making metastasis a late event in tumorigenesis.
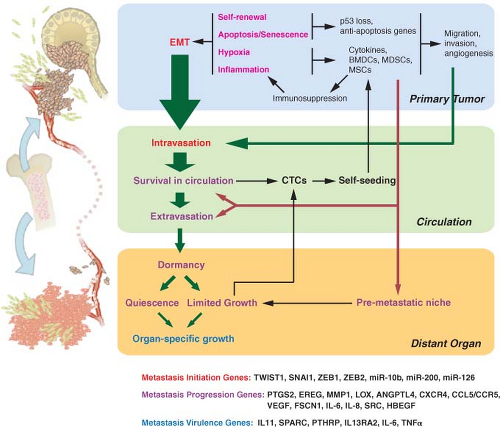 FIGURE 10.1 Selective pressures and steps from primary tumor growth to metastasis. Selective pressures at the primary tumor (labeled in magenta) can determine metastatic potential. Cancer is initiated by oncogenic changes. Of particular relevance to metastatic potential may be self-renewal pathways and the need to overcome apoptosis and senescence. Hypoxia and inflammation have important roles and lead to tumor cells co-opting bone marrow-derived cells (BMDCs), myeloid-derived suppressor cells (MDSCs), and mesenchymal stem cells (MSCs), to name a few. These cells and the cytokines that they produce enhance the ability of the tumor to migrate, invade, overcome hypoxia, and maintain an immunosuppressed environment. The ability of primary tumor cells to undergo an epithelial-to-mesenchymal transition (EMT) is also influenced by the selective pressures faced during primary tumor growth. EMT results in migration, invasion, and intravasation. Such functions (labeled in red) are examples of metastasis initiation functions. Although non-EMT-related migration and invasion can also lead to intravasation, EMT is likely a principle means by which circulating tumor cells (CTCs) are promoted. Further steps in the metastatic cascade are shown by green arrows, with the size of the arrow representing the likelihood that the step is successfully completed for many cancer types (e.g., breast cancer). Selective pressures encountered from the local microenvironment shape metastatic proclivity by selecting for tumorigenic functions that secondarily help cells navigate the metastatic cascade (metastasis-progression functions). Hypoxia and inflammation-related events contribute to metastasis-progression functions (labeled in purple) by enhancing the ability of CTCs to survive in the circulation, extravasate, and form a premetastatic niche. CTCs can either self-seed the primary tumor, which results in augmentation of tumor mass and further selection of metastatic traits, or extravasate into distant organs. Although a premetastatic niche facilitates adaptation to the foreign microenvironment of distant organs, further selection is needed for full colonization. This results in a period of dormancy whereby micrometastases remain quiescent or growth is counterbalanced by apoptosis and the lack of angiogenesis. Further somatic evolution within the distant organ can eventually result in selection of macrometastatic-colonization functions (labeled in blue) and organ-specific growth. On the bottom of the figure are examples of specific genes that play a role in metastasis initiation, metastasis progression, and macrometastatic colonization. |
hypoxic cells adapt. Up-regulated angiogenesis genes include vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF). These factors cause quiescent blood vessels to undergo remodeling, including the laying down of a matrix that activated endothelial cells use to form newly vascularized areas. Various glycolysis genes are expressed and their metabolic by-products lead to acidification of the extracellular space. This is normally toxic to cells and requires further adaptation either by up-regulation of H+ transporters or acquired resistance to apoptosis. To assist in invasion toward newly vascularized areas, HIF-α upregulates matrix metalloproteinase 1 and 2 (MMP1, MMP2), lysyl oxidase (LOX), and the chemokine receptor CXCR4. Degradation of the basement membrane by MMP2 and alteration of the extracellular matrix (ECM) by MMP1 and LOX clears away a barrier to migration. The activation of CXCR4 then stimulates cancer cells to migrate to regions of angiogenesis. Thus, if these series of events can be successfully completed, not only will preinvasive tumors successfully deal with hypoxia, but they will also likely invade through the basement membrane in the process. Invasion through the basement membrane defines invasive carcinomas.
activate fibroblasts. These activated fibroblasts become carcinoma-associated fibroblasts (CAFs) and promote primary tumor growth by secreting CXCL12 to stimulate CXCR4 on tumor cells. Angiogenesis is also aided by the action of CAFs through recruitment of endothelial progenitor cells by CXCL12 and by the action of TAMs that are recruited to areas of hypoxia to produce VEGF. Figure 10.2 presents a summary of this interaction. Thus, although the question of whether the immune system is a friend or foe of malignancies is not a new one, it would seem that recent answers suggest that cancers actually find ways to turn an enemy into an accomplice.
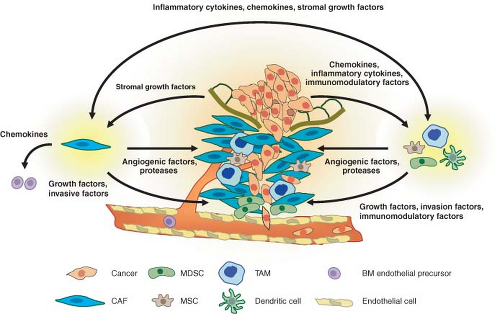 FIGURE 10.2 Interactions between cancer and stroma that promote invasion and metastasis. Cancerized stroma consists of fibroblasts, inflammatory cells, and other bone marrow-derived cells that have been conscripted to aid the tumor in overcoming hypoxia and in invasion and migration. Tissue breakdown, hypoxia, and inflammatory cytokines and chemokines secreted by the tumor cells result in recruitment of tumor-associated macrophages (TAMs), carcinoma-associated fibroblasts (CAFs), mesenchymal stem cells (MSCs), and myeloid-derived suppressor cells (MDSCs). TAMs and MDSCs can be found at points of basement membrane breakdown and at the invasive front of the tumor. These cells produce angiogenic factors to promote vascularization, proteases to degrade the extracellular matrix, and growth factors that stimulate tumor invasion and motility. CAFs also produce similar angiogenic factors, protease, and tumor growth factors. In addition, CAFs recruit bone marrow-derived endothelial precursors for angiogenesis. The cytokines and growth factors that TAMs and CAFs secrete are mutually beneficial to each other as part of an inflammatory/woundlike response. Cancers have been described as “wounds that do not heal.” This chronic state is maintained by immunomodulatory cytokines that suppress immune functions to ensure a protumorigenic environment. |
by immune cells for elimination. Cancer cells invariably ignore these cues, and their ability to resist cell death likely contributes to successful establishment of tumors.
and, for convenience, the collective denomination of EMT. Hypoxia can induce Snail and Twist, a direct target of HIF-1α.34 Low oxygen enhances β-catenin activity by inhibiting the activity of glycogen synthase kinase-3β, which normally induces the destruction of β-catenin. Accordingly, the presence of enhanced β-catenin signaling promotes Snail expression and subsequent EMT. Interestingly, the ability of hypoxia to liberate active β-catenin may set in place a feed-forward loop to help maintain EMT. Activation of Snail represses E-cadherin, which can then further enhance β-catenin and reinforce Snail expression.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


