The primary function of a clinical laboratory is to provide accurate and clinically relevant data for the diagnosis of medical conditions in patients. Laboratory data can also be used to provide patients with a management plan that will help them achieve a desirable outcome. Errors in the laboratory test results affect patient care when they mislead clinicians into false diagnoses or indicate a test is normal when it is not. With regard to laboratory tests, there are three potential sources of error: errors may occur before testing begins (preanalytical errors), during the test procedure (analytical errors), and following completion of the test (postanalytical errors).
Although errors can occur in any laboratory test result, immunoassays seem to be particularly vulnerable to analytical errors, due to the nature of antibody–antigen interactions and the potential for interference from cross-reactive species. By their very nature, antibodies have high but not absolute specificity for the antigen they are intended to detect, and therefore detection of known or unknown antigens with which the antibody reacts limits the specificity of immunoassays. In addition, reagent antibodies can also be antigenic, causing false-positive or false-negative signals due to the reaction of endogenous antibodies with the reagent. Although the frequency of medically important immunoassay interference is probably less than a few percent when it occurs, it can often lead to unnecessary treatment or undetected disease. Many of the analytes measured by immunoassays, such as tumor markers, cannot easily or cost-effectively be corroborated by other laboratory tests. Immunoassay interference, therefore, can lead to misinterpretation of patient’s status, misdiagnosis, unnecessary and costly radiological procedures, and/or unnecessary treatment.
Immunoassay interferences can originate from either exogenous or endogenous sources. Exogenous interferences are caused by the addition of external factors or conditions either in vivo or in vitro that are not normally present in a native, properly collected, and stored sample . For example, hemolysis, lipemia, blood collection tube additives, and administration of radioactive or fluorescent compounds, drugs, herbal medicines, nutritional supplements, and sample storage are all exogenous interferences that can adversely affect immunoassays . Interference from exogenous factors is an often-overlooked problem in immunoassays.
Endogenous interferences are caused by factors that may exist in the patient’s blood under either physiological or pathophysiological conditions . Endogenous factors often are difficult to detect and eliminate because they can vary considerably between patients, and from time to time in any given patient. Examples of endogenous interferences include human antianimal, heterophilic antibodies, autoantibodies , rheumatoid factors, and binding proteins . Knowledge of the exogenous and endogenous interferences and the mechanisms through which they interfere with immunoassays is essential to minimize the potential for analytical errors.
Although much of this chapter will be devoted to sources of error in immunoassay, because it is the most common analytical method used for measurement of hormones, many of the mechanisms by which endogenous and exogenous compounds interfere with immunoassays are analogous to interferences in chemical tests. The laboratory evaluation of endocrine disorders is not limited to immunoassays, and in this handbook, you will find many references to laboratory tests that do not involve antibodies. However, more considerable attention is given to immunoassays because they are commonly used in this domain and are uniquely susceptible to some of the interferences discussed in this book.
Interferences associated with mass spectrometry
Beginning in the early 2000s, the use of high-performance liquid chromatography (LC) with tandem mass spectrometric detection has transformed and vastly improved the analysis of small molecules, including steroid hormones. Beginning in 2010, the Centers for Disease Control (CDC) has worked to standardize steroid hormone analysis with their Hormone Standardization (HoST) project and Vitamin D Standardization Project (VDSP) . In 2013 the endocrine society began requiring mass spectrometry (MS) for sex steroid assays for publication in the Journal of Clinical Endocrinology and Metabolism . The use of MS or more often LC-MS/MS has historically been laboratory-developed tests; however, in 2017 the first LC-MS/MS test for 25-hydroxyvitamin D was cleared by Food and Drug Administration (FDA) . The LC-MS/MS platform, owing to its core technology and granularity of data collection, is widely considered more specific, more selective, and less prone to interference than most immunoassays for small molecule (steroid) analysis. There are, however, some particular areas that laboratorians must address during validation of LC-MS/MS assays . When choosing a laboratory and diagnostic test, it is important to work with a laboratory that is experienced, accredited, using modern tools, and validating to current expectations based on Clinical Laboratory Standards Institute (CLSI) guidelines including C50-A and C62 . In addition to steroid analysis, MS is now being used for peptide and protein analysis within the endocrine space for selected analytes, including insulin-like growth factor 1, thyroglobulin, and plasma renin activity (angiotensin I). Protein analysis by MS is more challenging than the analysis of small molecules but offers extraordinary potential for biomarker analysis.
Exogenous interference
Hemolysis, lipemia, and icterus
Hemolysis, icterus, and lipemia can interfere with many laboratory tests. Hemolysis can affect the absorbance of light in spectrophotometric measurements since hemoglobin is a chromophore that absorbs broadly in the ultraviolet (UV)-visible range of the electromagnetic spectrum. Hemolysis occurs when erythrocytes are disrupted; the normal concentration of hemoglobin in the plasma is very low. When erythrocytes are disrupted, their cytoplasmic contents add soluble factors to the plasma fraction that remain after centrifugation. These soluble factors may bind to the analyte and block antibody-binding sites or cross-react with the reagent antibodies. Hemolyzed specimens may be unacceptable for immunoassays of labile analytes such as insulin, glucagon, calcitonin, parathyroid hormone (PTH), adrenocorticotropic hormone (ACTH), and gastrin because proteolytic enzymes released from erythrocytes can degrade peptide hormones . Hemolysis may also interfere with signal generation steps included in various immunoassays . Hemolysis may interfere with spectrophotometric measurements due to absorption by hemoglobin; this is a hemolysis interference that is not specific to immunoassays. Finally, for some analytes, the intracellular concentration is much greater than the extracellular concentration; therefore disruption of the erythrocytes increases the analyte concentration in the plasma above its physiological level. In general, grossly hemolyzed specimens should not be used for either chemical assays or immunoassays.
Lipemia can result from high concentrations of triglycerides, cholesterol, or both, and may produce erroneous results in some assays by interfering with antigen binding, even when antibodies are linked to a solid support. Lipemia has also been shown to cause interference with immunoassays that have turbidimetric end-points because of the light scatter caused by lipid micelles . Interferences by nonesterified fatty acids have been well documented for free thyroxine assays. Nonesterified fatty acids compete with thyroxine and its derivatives used as labels for endogenous protein binding sites and, depending on the assay format, may cause either falsely high or falsely low free thyroxine values . Nonesterified fatty acids may also inhibit steroid binding to proteins. Hypertriglyceridemia has been shown to cause falsely elevated results in some endocrinology assays, using second antibody and polyethylene glycol separation techniques . It is desirable to collect specimens from individuals after an overnight fast to reduce the immunoassay interference from lipids. Alternatively, ultracentrifugation to remove any excess lipids or enzymatic cleavage by lipase may be used to treat samples before analysis .
It should also be kept in mind that blood contains three distinct fractions: a cellular fraction that includes erythrocytes, leukocytes, and platelets; an aqueous fraction that includes primarily water and various small molecules, including peptides; and an extracellular nonaqueous fraction that consists of lipid micelles and globular proteins. The cellular fraction comprises approximately 45% of the total blood volume. When a specimen is centrifuged, the noncellular fraction is usually about 95% aqueous, with the remaining 5% comprised of lipid micelles (such as lipoproteins) and globular proteins, including immunoglobulins. Therefore any analyte that is exclusively in the aqueous fraction of blood (electrolytes, organic acids, catecholamines, unbound steroid hormones) is only occupying around 95% of the total volume of plasma. The excluded nonaqueous volume introduces an error when concentrations are based on the total volume of the specimen. For most assays, this small error is inconsequential. However, in hyperlipidemic or hyperproteinemic states, the nonaqueous volume can be 10%–15%, which can cause a significant error in the calculation of analyte concentrations. For electrolytes, direct potentiometry solves this problem because measurements made in undiluted specimens are volume-independent—that is, the volume occupied by nonaqueous components in plasma is not relevant. However, there are no direct potentiometric methods for measuring hormones, and it is another reason why hyperlipidemic specimens should not be used for laboratory tests unless the lipids are removed by ultracentrifugation.
Excess bilirubin affects many different types of assays, either due to spectrophotometric interference or chemical oxidation of the analyte by bilirubin. As with the case for hemolysis and lipemia, the user should follow the recommendations of the manufacturer to determine the suitability of icteric samples for analysis.
Matrix effects
Clinical specimens are incredibly complex and contain a variable mixture of proteins, carbohydrates, lipids, small molecules, and salts. A matrix effect is an interference arising from any of the above substances present at high enough concentration to affect the measurement of an analyte. In addition, the viscosity, pH, and ionic strength of the specimen may also contribute to matrix effects. Matrix interference in immunoassays has been thoroughly investigated and reviewed . Antigen and antibody reactions are often quite sensitive to variations in protein and lipid concentrations, pH, and ionic strength. Differences in the matrix may alter the efficiency of separation of bound and unbound fractions and the extent of nonspecific binding of the tracer. Matrix effects are highly dependent on the assay format and the antibody selected to capture analyte; therefore various immunoassays are affected in different ways. Matrix effects can be important sources of discrepancies among patient samples, calibrators, standards, quality controls, and external proficiency materials that are not based on human serum .
Additionally, matrix effects can make certain types of body fluids unsuitable for analysis . Thus matrix interferences in clinical assays can be minimized by careful assay design. Examples of methods to reduce or eliminate matrix interferences include the use of high-affinity antibodies of certain subtypes or antibody fragments, dilution of the specimen, addition of an immunoglobulin to the assay buffer to saturate any interfering antibodies, and optimizing temperature and incubation times. When it is necessary to dilute a sample with a high concentration beyond the linear range of the assay, it is important to use a diluent recommended by the manufacturer with the proper matrix to avoid any artifacts.
Blood collection tubes
Blood collection tubes are not inert containers for blood but have several constituents, including anticoagulants, surfactants, and lubricants for rubber stoppers, clot activators, and separator gels that can potentially interfere with the assays.
Tube wall
Serum separator tubes and Vacuette tubes are made from polyethylene terephthalate. Siliconized plastic tubes have been shown to cause a 30%–60% decrease in corticotropin (ACTH) measurements when measured by radioimmunoassay, possibly due to interference with the formation of either the biotin-avidin complex or the antibody–antigen–antibody sandwich . Besides tube walls, it is important to note that other synthetic materials that come in contact with blood, such as indwelling catheter tubing, have also been reported to affect immunoassay results . Certain drugs, such as tacrolimus, can tightly bind to the catheter tubing wall, resulting in spuriously elevated drug concentrations when blood is collected through catheters, even after flushing the line with saline .
Rubber stopper
To maintain the vacuum in blood collection tubes, it is common to use stoppers made from isobutylene-isopropene rubber or chlorinated isobutylene-isoprene rubber along with a stopper lubricant . Rubber stoppers used in some commercial blood collection tubes have been found to contain a plasticizer, tris(2-butoxyethyl) phosphate, which can displace quinidine, propanolol, lidocaine, tricyclic antidepressants, fluphenazine, and chlorpromazine from α1-acid glycoprotein . Displacement of these drugs from their binding protein results in the redistribution of the drug in blood, causing an increase in drug uptake by red blood cells and a decrease in plasma or serum concentration . Most manufacturers have reformulated their rubber stoppers with low extractable rubber to minimize this interference.
Anticoagulants
Historically, most assays were performed in serum, but plasma may be preferable because it eliminates the time required for clotting, thereby reducing the processing time . If plasma is used, care must be taken to select an anticoagulant that does not interfere with the assay.
Ethylenediaminetetraacetic acid (EDTA) binds divalent metal ions and prevents coagulation by binding to calcium. EDTA also binds some metal ions that are a constituent of labels: for example, europium, or enzyme cofactors essential for their activity (e.g., alkaline phosphatase requires zinc ions) . Elevated EDTA concentrations in a sample-reagent mixture due to insufficient sample volume can result in the more efficient chelation of magnesium and zinc and can affect the activity of the alkaline phosphatase enzyme label used in chemiluminescence assays . Filling of EDTA sample tubes to <50% affects, for example, intact PTH and ACTH measurements by the Immulite assays . Many proteins bind divalent cations—typically calcium or magnesium—and the antibodies against these proteins may recognize an epitope that is altered when the cation is absent .
Heparin prevents coagulation primarily by forming a complex with antithrombin III. The heparin-antithrombin III complex enhances the inhibitory effects of thrombin and activated Factor X to prevent clotting or activation of thrombin, which in turn prevents the formation of fibrin from fibrinogen. However, heparin may interfere with some antibody–antigen reactions. Heparin decreases the rate of reaction of some antibodies, particularly at the precipitation step in second-antibody systems; however, the use of solid-phase systems has minimized this problem. Heparin can precipitate cryofibrinogen; therefore this anticoagulant should not be used for cryoprotein measurements. The influence of exogenously administered heparin on serum levels of thyroid hormones and other analytes has also been investigated. Heparin has also been shown to cause in vivo stimulation of lipoprotein lipase with subsequent release of nonesterified fatty acids. Nonesterified fatty acids inhibited the binding of radiolabeled thyroxine to thyroid-binding globulin with an apparent increase in the thyroxine result. Samples collected into tubes containing sodium fluoride may be unsuitable for some enzymatic immunoassays due to inhibition of the enzyme activity by fluoride.
Surfactants
Surfactants are commonly added to immunoassay reagents to decrease or eliminate nonspecific adsorption of the analyte, improve reagent stability, or modify the solid-phase surface to render it less hydrophobic, thus minimizing the loss of noncovalently bound antibody . At high concentration, surfactants may inhibit passive adsorption of antibody from the solid phase, among other nonspecific effects. Previous reports have shown that silicone-coated collection tubes can interfere with ion-specific electrode determinations of ionized magnesium and lithium , causing falsely increased concentrations. In addition, the water-soluble silicone polymer coating the interior of serum separator tubes can interfere with the formation of an avidin-biotin complex in immunoradiometric assays for thyrotropin, prolactin, and human chorionic gonadotropin (hCG) . Bowen et al. identified a common organosilane surfactant (Silwet L-720) in Becton Dickinson SST blood collection tubes that caused a falsely elevated triiodothyronine and other analytes. When present in excess amounts in blood collection tubes, this surfactant causes interferences by desorbing the capture antibody from the solid phase used in the Immulite 2000/2500 triiodothyronine immunoassay and had a similar effect on immunoassays from other manufacturers . Other studies have shown that silicone forms a complex with C-reactive protein, enhancing the antigen–antibody reaction in the Vitros C-reactive protein assay and falsely elevating results .
Clot activators
Blood collected in evacuated tubes without anticoagulants should form a dense clot as rapidly and completely as possible to enable clear separation of the clot from the serum layer by centrifugation . To achieve this end, blood collection tubes include a clot activator with a carrier such as polyvinylpyrrolidone . Examples of clot activators include diatomaceous earth, particles of inorganic silicates, and biochemicals such as ellagic acid, thrombin, and thromboplastin. Occasionally, clot activator particles may not pellet completely with the clot and instead remain in the serum layer, causing interferences with some assays.
Separator gel
Separator gels are widely used in blood collection tubes to form a barrier between serum or plasma and the cellular fraction upon centrifugation. The separator gel is a thixotropic liquid that is solid at rest, but when compressed by centrifugation, it becomes liquid . It has been shown that the separator gel can adsorb some hydrophobic leading to falsely low results . Fragments of separator gel or droplets of oil may be seen within the separated serum or plasma following the centrifugation of some gel-containing blood collection tubes. The gel or oil droplets can clog the sample probe, coat tubes, and cuvettes, and physically interfere with solid-phase immunoassay systems. To minimize interferences, it is important to follow the tube manufacturer’s recommendations and not use the tubes at temperatures, centrifugation speeds, or orientations that are not within limits specified by the manufacturer.
Label interferences
All immunoassay requires a measurable indicator to quantify the antigen–antibody complex. Some samples may contain compounds that increase or decrease the magnitude of the indicator response without affecting antigen–antibody binding. Diagnostic or therapeutic administration of radioisotopes has the potential to interfere with radioimmunoassays if the same isotope is used as the indicator . When the indicator is a fluorophore, interference can occur due to endogenous fluorescence, fluorescent drugs, or fluorescein administered for retinal angiography . Fluorescence quenchers may also be present in some clinical specimens. For enzyme-labeled immunoassays, the presence of enzyme inhibitors or activators in the sample may alter the enzyme activity, and thus the results from the immunoassay .
Drugs and herbal medicine
A major source of exogenous interference in immunoassays is the presence of drugs, nutritional supplements, and herbal medicine in the blood. These interferences have been widely reviewed . Many of the interferences involve cross-reactivity of exogenous compounds with the capture antibody. Cross-reacting substances may be a precursor of the compound to be measured, or it can be the metabolite of the analyte.
Timing of sample collection
Although not strictly an interference, it is important to realize that some analytes have a marked diurnal pattern of physiological release and/or can be affected by variable physiologic factors or therapies. As a result, these analytes must be measured at specific times to ensure that results can be interpreted appropriately. Cortisol concentration, for example, varies significantly from a peak near midnight to a nadir at 7–8 a.m., and reference ranges are applied accordingly .
Storage/freeze-thaw
Inappropriate sample type, specimen processing, and storage can change the properties of the sample over time and affect immunoassay results. For example, ACTH is only stable for 18 h at 4°C, in contrast to 18 other hormones that are stable for >120 h . Most hormones are relatively stable in serum or urine if they are rapidly frozen and stored at −70°C; however, repeated freezing and thawing of analytes can lead to denaturation, aggregation, and loss of antigenicity of some proteins . Specimens collected in EDTA often are more stable than serum or heparinized plasma specimens because EDTA chelates calcium and magnesium ions, which are cofactors for some protease enzymes. The addition of protease inhibitors (e.g., aprotinin) to blood samples may also improve analyte stability .
Analytical errors
Carryover
Automated chemistry and immunoassay systems use automated sample-handling devices. If a sample to be assayed is preceded by a sample with a very high concentration of an analyte, there is a potential for the analyte in the first sample to contaminate the second sample due to inadequate rinsing of the probe between specimens. Therefore it is important to routinely test any new analyzer for potential carryover. It is the responsibility of the laboratory to assess the potential for carryover in each assay it performs and incorporate the appropriate action to take in the standard operating procedure.
Endogenous interference
Excess antigen interference (hook effect)
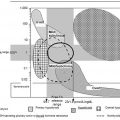
Excess antigen interference occurs when there is an unusually high concentration of antigen, which paradoxically may lead to a falsely low result. This phenomenon is commonly referred to as a “hook” or prozone effect . This type of interference is most likely to occur when the analyte can have a very wide range of concentrations, as do hCG, many tumor markers, and serological tests . As shown in Fig. 1.1 , 2-site immunometric assays are especially prone to the hook effect. These assays depend upon the formation of a complex between the antigen and the primary (or “capture”) and secondary antibodies to generate a signal. These assays are usually designed so that there are always excess antibodies relative to the antigen. When there is an excess antigen, however, a complex does not form because all of the antigen-binding sites are occupied by a single antigen, thus preventing a single antigen molecule from bridging the primary and secondary antibodies . After a final wash step, the secondary antibodies that generate the signal are removed when there is an excess antigen, thus resulting in a spuriously low value.
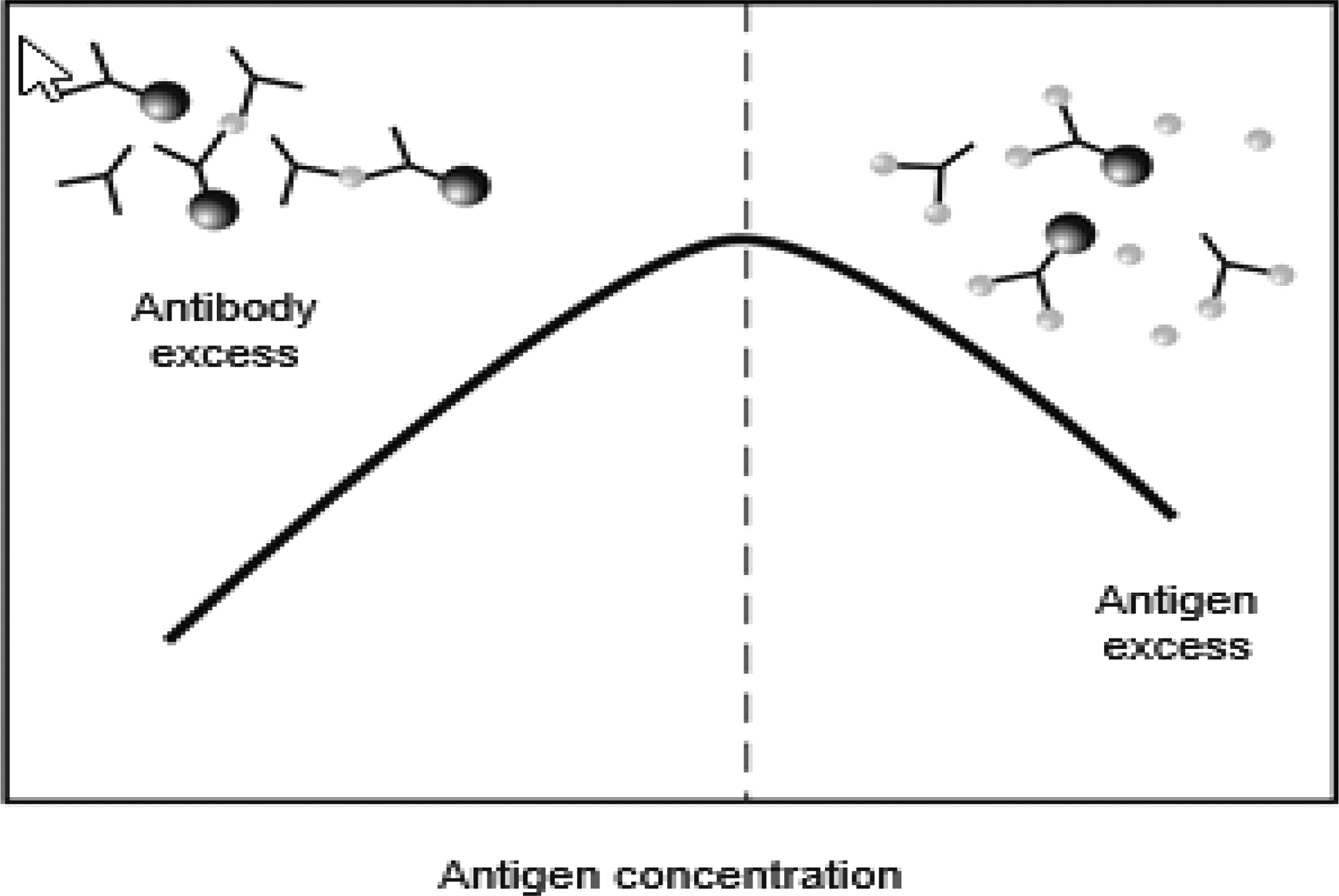
Antigen excess interference can be prevented by performing the assay on a diluted sample or by the addition of a vast excess of antibody to ensure that there is always a sufficient concentration of antibodies, even for those samples with unusually high concentrations of the analyte. Manufacturers of assays for analytes that are commonly affected by antigen excess usually provide a sufficient amount of antibodies to prevent this problem. In addition, sequential two-step immunometric assays are less prone to this problem because a wash step is included before the addition of the second antibody; this wash removes any excess antigen, preventing an erroneously low result. Finally, an important consideration in implementing any new assay that may be susceptible to the hook effect is to determine whether the linear range described by the manufacturer encompasses the likely concentration range that will be measured in the laboratory.
Antibody specificity (cross-reactivity)
Specificity is an important characteristic of any assay because it describes its ability to react with only the analyte of interest in a sample. A major source of bias in an immunoassay is the lack of specificity of the antibodies used, resulting in cross-reactivity. Cross-reactivity is more problematic with polyclonal antibodies compared to monoclonal antibodies. Polyclonal antibodies typically have high avidity, but monoclonal antibodies have a higher affinity and therefore are more selective toward a specific antigen. Selection of monoclonal antibodies may reduce cross-reactivity, but closely related molecules may still be recognized by the antibody, especially analytes, such as steroids and drugs. Cross-reactivity is more common with competitive immunoassays compared to noncompetitive immunoassays since the use of an antibody against a second epitope on the analyte reduces the likelihood of a cross-reacting antigen having both epitopes tested for by the noncompetitive immunoassays. However, noncompetitive immunoassays do not always exhibit good specificity because nonspecific binding may arise due to many endogenous substances present in the patient sample, such as rheumatoid factor, complement proteins, or bacterial proteins .
Antibody interference
Circulating antibodies in patient samples may interfere with immunoassays . Usually, endogenous antibodies interfere by reacting with the reagent antibodies in immunoassays, but all the other components of immunoassays, such as the antigen, enzyme-substrate, and signal molecule, can also be a target of endogenous antibodies, adversely affecting immunoassay results. Previous studies have shown that interfering endogenous antibodies are present in up to 30%–40% of patient samples , but estimates vary widely , and their practical effect is far less. Interfering antibodies include heterophilic antibodies that have multiple specificities for antigens, as well as proteins, such as rheumatoid factor; human antianimal antibodies (HAAAs), especially human antimouse antibodies (HAMAs); or autoantibodies. The presence of interfering antibodies can affect both noncompetitive and competitive immunoassays and produce either falsely elevated or falsely decreased results, depending on the specific antibody or protein with which the heterophile reacts. The nature of the antibody interference depends on the type of assay used and the site where the antibody binds to the analyte .
Heterophile antibodies
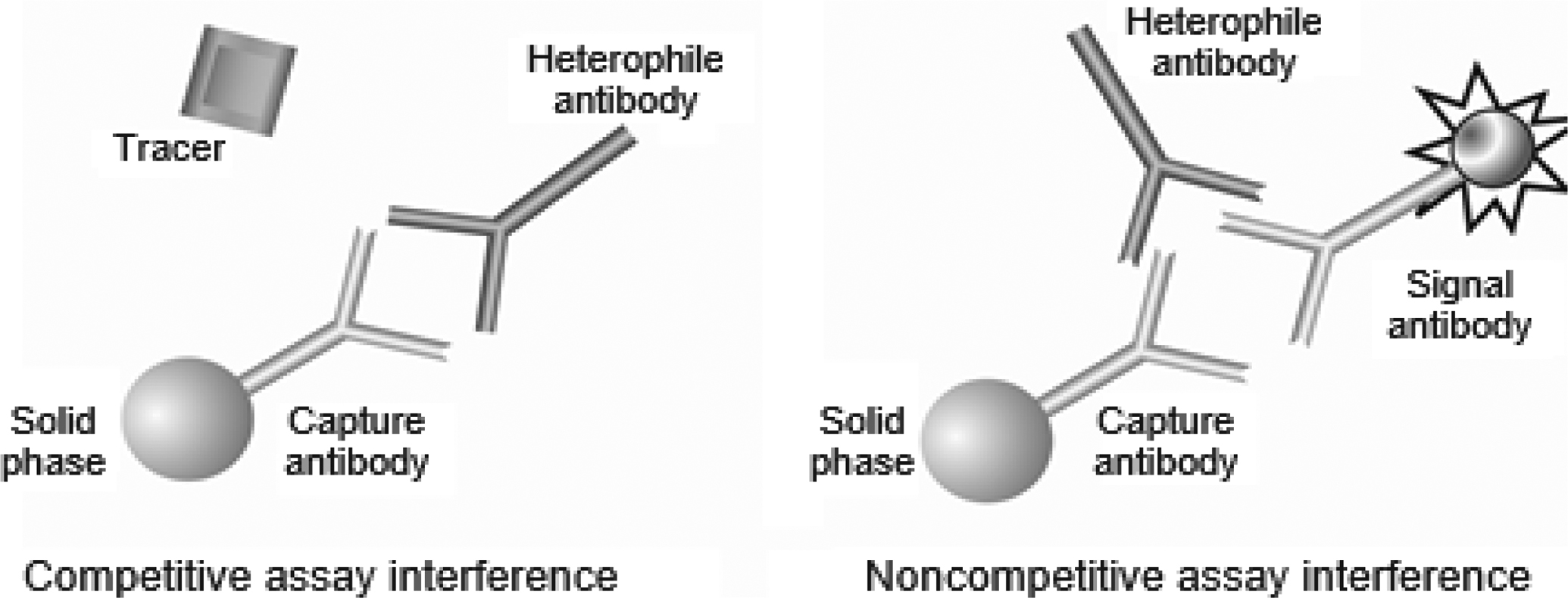
Heterophile antibodies can be immunoglobulin G (IgG), immunoglobulin M (IgM), immunoglobulin A, and IgG isotypes that are poorly defined and react with a wide spectrum of antigens . If exposure to a specific animal immunogen is known, the correct term for that heterophile antibody should refer to the specific animal that produced the antigen such as “human antimouse antibody,” or “human antirabbit antibody.” Heterophile antibodies are usually low-titer, weak-avidity antibodies found in the serum of patients with no history of treatment or diagnostic procedures involving antianimal immunoglobulins . Heterophile antibodies have been reported to be present in 30%–40% of patient samples and are a well-recognized cause of interference in immunoassays . When heterophile antibodies are present, it is often very difficult to predict the direction and magnitude of the immunoassay interference. Interferences may occur in competitive or noncompetitive assays, but the latter is more common . In competitive immunoassays, the presence of heterophile antibodies may decrease the number of available binding sites on the primary antibody by steric hindrance of the specific antigen, ( Fig. 1.2 ) and in some competitive immunoassay formats, reduce the binding of a second antibody, causing a positive interference. In noncompetitive immunoassays, heterophile antibodies can interfere by bridging the capture and detection antibodies , causing false-positive results ( Fig. 1.2 ). In contrast, heterophile antibodies can also cause false-negative results by binding directly to the capture antibody, thus blocking the reactive site from binding the analyte of interest. The same antibody may react differently for different antibody combinations, thus causing falsely elevated results in one assay but a lower result in another assay. Immunoassay manufacturers typically add blocking agents (nonimmune globulins of various species) to their assay formulations to saturate and minimize or eliminate the effects of heterophile antibodies; however, not all heterophile interference can be prevented by blocking agents . Heterophile antibodies may show reactivity to idiotypes that are not present in the blocking agent . Prior extraction of the analyte from the sample by protein A, protein G, cation-exchange, or gel filtration chromatography is also effective in removing heterophile antibodies . Reaction conditions can also be modified to minimize heterophile antibody interference Heat and acid treatment of samples are not very useful since these antibody-denaturing conditions will destroy most analytes.
Human antianimal antibodies
HAAAs are high-affinity, specific polyclonal antibodies that react with immunoglobulins from a specific animal. For antianimal antibodies elicited by animal immunoglobulins, the HAAAs can have antiidiotype or antiisotype specificity. Antiidiotype antibodies bind to the hypervariable region of the immunoglobulin molecule, and antiisotype antibodies are directed against the constant regions of the immunoglobulin molecule. Anti-antiidiotype antibodies can also be produced; these antibodies recognize the binding region of the antiidiotype antibody; thus the antigen-binding region of an anti-antiidiotype antibody resembles the antigen that elicited the original antiidiotype HAMA. The prevalence of HAAAs in serum samples has been shown to vary widely among studies with ranges anywhere from 0.1% to 80%, depending on the assay used to detect them and the patient population . HAAAs may be produced from iatrogenic and noniatrogenic causes, including the use of mouse monoclonal antibodies for therapeutic and imaging purposes, blood transfusions, vaccinations, exposure to microbial antigens, animal husbandry or keeping animals as pets, transferring of a dietary antigen across the gut wall, and autoimmune diseases that give rise to autoantibodies. Antianimal antibodies are known to interfere unpredictably with immunoassays, the effect on laboratory results depending on the nature and concentration of the interfering antibody, and the immunoassay format. HAAAs most commonly interfere with 2-site immunometric assays by linking the capture and detection antibodies, giving a false-positive result .
Human antimouse antibodies
HAMAs are heterophile antibodies that react with mouse immunoglobulins . Many cases of immunoassay interference due to HAMAs have been reported and reviewed. In one study evaluating the incidence of HAMAs, eight commonly used immunoassays were tested in 500 serum samples . It was found that the incidence of HAMAs ranged from 0.2% to 3.7%. The incidence of HAMAs has increased since the introduction of in vivo techniques using mouse monoclonal antibodies as carriers for radiological or chemotherapeutic agents to tumor sites and for other therapeutic and imaging purposes. Two-site immunometric methods are more susceptible to interference from antibodies to animal IgG in human serum and may cross-react with reagent antibodies to animal IgG in human serum, as well as with reagent antibodies, especially those from the same species. Reaction with both capture and detection antibodies forms an antigen–antibody complex that behaves immunochemically like the analyte-antibody complex, resulting in false-positive results. False-negative results can also occur in 2-site immunometric assays due to HAMAs reacting with one of the reagent antibodies and blocking the formation of the antigen–antibody complexes with the analyte of interest. Methods that use only one mouse monoclonal antibody in immunometric assays are less prone to interference from HAMAs.
Autoantibodies
Autoantibodies are antibodies that exist in a patient’s blood that bind preferentially to the analyte of interest. Autoantibody prevalence is between 4% and 27% in the normal population. Endogenous circulating autoantibodies have been described for a variety of analytes, including thyroid hormones, thyroxine, and triiodothyronine, as well as thyroglobulin , creatine kinase, amylase, insulin , prolactin , and testosterone . The nature of autoantibody interference on immunoassay results depends on the assay technique used as well as the autoantibody titer, affinity, and specificity . In competitive immunoassays, autoantibody interference can lead to an apparent increase or decrease in measured concentration, depending on whether the autoantibody-analyte complex partitions into the free or bound fraction. In noncompetitive immunoassays, autoantibodies can bind to the solid support or block attachment of detection antibody and cause falsely low results by decreasing the amount of label bound to the solid-phase antibody. Besides interfering with immunoassays, autoantibodies against an analyte can delay its in vivo clearance, resulting in a true serum elevation. This is commonly encountered with prolactin, which can lead to a diagnosis of a pituitary adenoma, but the prolactin bound to the autoantibody is not biologically active and does not lead to disease . Autoantibodies to reagent components, such as alkaline phosphatase, may also yield incorrect results for reagents using that enzyme . Thus regardless of whether competitive or noncompetitive immunoassays are used, interference from autoantibodies can occur in both formats.
Rheumatoid factors
Rheumatoid factors are IgM autoantibodies directed against the Fc portion of IgG . These factors bind to a hydrophobic domain on the antibody that is close to the binding site of staphylococcal protein A . It has been estimated that ~5% of healthy individuals have positive rheumatoid factors . Positive rheumatoid factors have been found in varying percentages in other connective tissue disorders, such as systemic lupus erythematosus, systemic sclerosis, polymyositis, and Sjögren syndrome. Rheumatoid factors can interfere with immunoassays by binding to the Fc portion of immunoglobulins and causing steric hindrance to the binding site, preventing or decreasing binding to the antigen or by cross-linking the reagent antibody, thereby mimicking the antigen. Monoclonal antibody-based immunometric assays are especially sensitive to the presence of heterophile antibodies . The nonspecific binding by rheumatoid factor can be overcome in the same manner as for heterophile antibodies by blocking reagents such as nonimmune homologous immunoglobulin .
Endogenous hormone-binding proteins
Endogenous hormone-binding proteins are present in varying concentrations in all serum and plasma samples and may significantly affect immunoassay performance. Immunoassays may also be affected by changes in specific binding proteins due to congenital variations; for example, in familial dysalbuminemic hyperthyroxinemia, up to 50% of the albumin molecules bind thyroxine with an affinity up to 50-fold higher than normal. Measurement of total hormone concentration requires the displacement of the hormone from its binding site. Unless the protein binding of both labeled and unlabeled ligands is inhibited or removed, equilibrium will form among the binding proteins, the labeled and unlabeled ligands, and the analytical antibody, producing erroneous results. Therefore total hormone assays need to remove the endogenous binding protein to prevent the binding of the added signal-ligand complex to the endogenous binding sites. Various releasing agents have been used to disassociate the hormones from the binding protein, such as pH changes, 8-anilino-1-naphthalenesulfonic acid, salicylate, and thimerosal.
Other plasma proteins
Other plasma proteins that can affect antibody binding and interfere in immunoassay include fibrin, complement, lysozyme, paraprotein, and albumin.
Fibrinogen
Immunoassays are susceptible to interference by fibrin. Residual fibrin may be present because of improper specimen handling procedures. It can be present in the primary tube samples either as a visible clot, which may physically occlude the sample probe on the automated chemistry or immunoassay instrument, or more insidiously, as an invisible microfiber or as strands. Fibrin interference is usually not reproducible and can disappear with time as the fibrin settles out of the sample. Although newer immunoassay analyzers often have clot detectors, they are not infallible, and it is still common laboratory practice to repeat the analysis for those samples with an analyte level below the detection limit to minimize this type of problem. Excess fibrin can also have specific deleterious effects on immunoassays. Some immunoassays are directly affected by fibrin interference , especially with high surface–area solid supports, presumably caused by nonspecific binding of a conjugate molecule to fibrin or other coagulation intermediates deposited on the solid surface.
Complement proteins
Many immunoassays use antibodies bound to a solid phase. When a serum sample is added to the immobilized reagent antibodies, the various complement factors in the samples are activated and bind to the reagent antibodies . This binding may partly or totally block the antigen-binding sites, and it has been shown that complement activation may interfere with antigen binding to the capture antibody and cause falsely negative results. Complement binding may also lead to the destruction of the antigen–antibody complexes. Mouse immunoglobulin IgG2a and IgG2b subtypes are particularly sensitive to complement binding and activation. The effect of complement can be eliminated by diluting or heating the samples or by using EDTA-containing buffers, which chelate calcium and inactivate complement . In addition, since complement can interfere with immunoassay by binding to the Fc fragment of immunoglobulins, it is advantageous to use F(ab) fragments that are resistant to complement binding .
Lysozymes
Lysozyme is a basic protein with an isoelectric and isoionic pH of 11.1. It is widely distributed and possesses bacteriolytic properties. As a highly basic protein, lysozymes can bind anionic proteins with low isoelectric points like immunoglobulins (pI values between 5.0 and 9.0) . Lysozymes also nonspecifically bind to the immunoglobulin and can form a bridge between solid-phase IgG and detection antibody . The inclusion of copper ions or ovalbumin in some enzyme-labeled immunoassays has been shown to minimize lysozyme interference .
Paraproteins
Elevated concentrations of paraproteins due to multiple myeloma and similar diseases in serum may result in nonspecific binding of either analytes or reagents in immunoassays or interference in nephelometric assays . Paraproteins also greatly increase the viscosity of the serum, which can produce short samples and cause falsely low immunoassay values.
Albumin
Albumin may interfere with immunoassay results by binding, as well as releasing a large quantity of ligands. Methods measuring the unbound analog of free thyroxine may actually measure the albumin-bound hormone fraction and be influenced by the level of albumin in the serum. Interpretation of free thyroxine values should be evaluated with caution in patients with abnormal concentrations of serum albumin, for example, those with nonthyroid severe illness. In conditions associated with low or absent albumin (hereditary analbuminemia) or abnormal albumin binding of analog tracer (familial dysalbuminemic hyperthyroxinemia), false low or false high levels of free thyroxine, respectively, may be obtained. In some solid-phase assays, albumin may even inhibit the binding of labeled thyroxine to the immobilized thyroxine antibody, producing falsely high results. If a patient is on heparin therapy, the concentration of the nonesterified fatty acids is increased due to release of lipoprotein lipase from the vascular epithelium, and nonesterified fatty acids will bind strongly to albumin, displacing residual labeled thyroxine, the concentration of which increases in the specific bound fraction, resulting in apparently lower values for free thyroxine.
Detection and testing for interference in suspected samples
There is no universal way to detect all types of potential interference in chemical and immunoassays; therefore processes are needed to make both medical laboratory workers and physicians aware of the potential for interference. Once the laboratory is alerted to a potential problem, several laboratory procedures can be used for investigating these specimens. Additional studies to investigate potential interferences include: (1) retesting the same specimen, (2) testing a new specimen from the same patient, (3) testing another body fluid (e.g., urine hCG) to corroborate the result, (4) dilution studies to assess linearity, (5) spiked recovery, (6) mixing experiments with a normal serum or plasma sample, (7) heterophile antibody-blocking reagents; (8) sample pretreatment, and (9) testing with alternate technology.
Not all of these above procedures are needed or applicable to all situations, and frequently, repeat testing or testing on a new specimen will resolve a problem. If an interference is still suspected, dilution studies may confirm whether the diluted concentrations produce a linear signal response. Diluting the sample may lower the concentration of the interfering substance below the limit at which it interferes with the assay. Dilution studies should only be attempted with the diluent recommended by the manufacturer, and ideally, another specimen without a suspected interference should be diluted as a control. Another approach is to pretreat the sample with various commercial additives to neutralize any problems from antibody interference. Because many immunoassay interferences are specific to certain assays, it is also helpful to perform the assay using another method. Methods are available to detect heterophile antibodies, but often this is not useful because heterophile antibodies are relatively common and usually do not cause problems. In addition, assays to detect endogenous antibodies are usually very specific to a certain class of antibodies, and no single test is sufficient to exclude the presence of an interfering antibody. A very high titer of endogenous interfering antibodies may be a clue to the source of an immunoassay interference, but this can also be tested empirically by adding blocking agents to neutralize these antibodies. Rarely, interferences can result from an inherent and/or systematic limitation of a specific assay or blood collection tube. In these instances, it is important to notify not only the physicians ordering the test but also the manufacturer of the assay and any relevant regulatory agency, such as the FDA, of the interference.
Biotin
Biotin is a B vitamin found in small amounts in a variety of foods. Biotin also is sold in nail, hair, and skin supplements, and is prescribed in high doses for patients with neurological diseases such as progressive multiple sclerosis . Biotin-streptavidin coupling is frequently used in immunoassays to capture biotinylated antibody–antigen complexes . High concentrations of biotin can interfere with biotin-streptavidin-based immunoassays, producing erroneous test results . Although the concentrations of biotin necessary to interfere with an immunoassay are substantially higher than normal blood biotin levels, or those associated with ingesting 30 µg/day of biotin, the risk of biotin interference with immunoassays is increasing, as many contemporary supplements incorporate higher-dose biotin of up to 20 mg .
Prevalence of elevated biotin
Estimates of the prevalence of biotin supplement use in the United States range from 8% to 29% . However, as biotin is sold under various names, patients may not be aware of their biotin supplement use and may not report it to their health care provider . Efforts to quantify the prevalence of elevated biotin levels in the blood have produced inconsistent results. One study found ~0.74% of the routine samples assessed contained concentrations of >20 ng/mL , while another study found that ~ 7.4% of the samples contained concentrations of ≥10 ng/mL . Cardiac troponin assays have also been found to have a relatively low biotin interference threshold, which could result in a missed diagnosis of acute myocardial infarction .
Peak serum or plasma levels of biotin are achieved 1–2 h after ingestion and afterward decline quickly . Median serum peak concentrations in healthy volunteers were 41, 91, and 184 ng/mL 1 h after biotin consumption of 5, 10, and 20 mg, respectively; peak biotin concentrations following a 20 mg biotin dose did not exceed 355 ng/mL. The time required to achieve serum biotin concentrations of 10 and 30 ng/mL was 1.5–147 and 0–105 h, respectively, for individuals who received a daily dose of biotin from 1 to 300 mg. The maximum plasma biotin levels were shown to be <1200 ng/mL following a 300 mg biotin dose.
Impact of biotin interference on diagnostic assays
The risk of erroneously low (noncompetitive “sandwich” immunoassays) or high (competitive immunoassays) results varies according to the assay design, mechanism of interference, and the analyte and biotin concentrations in the sample . In noncompetitive immunoassays, the signal- and biotinylated antibodies bind the analyte, which subsequently captures the analyte-antibody sandwich complex on a streptavidin-coated solid phase. In the absence of interference, the signal rises as the analyte concentration increases. Excess biotin can saturate the streptavidin binding sites and prevent the capture of the analyte-antibody sandwich complex on the streptavidin-coated solid phase, causing erroneously low results ( Fig. 1.3 ). Labeled (signal) and endogenous analytes compete for biotinylated antibody linking sites in competitive immunoassays, and the biotinylated antibody is subsequently linked to the streptavidin-coated solid phase. In the absence of interference, the signal decreases as the analyte concentration increases. When biotin concentrations are high, excess biotin binds to the solid phase and prevents the antigen–antibody complexes from binding to the solid phase. Unbound antibodies are removed during washing, deceptively reducing the signal and generating inaccurately high assay results ( Fig. 1.3 ).
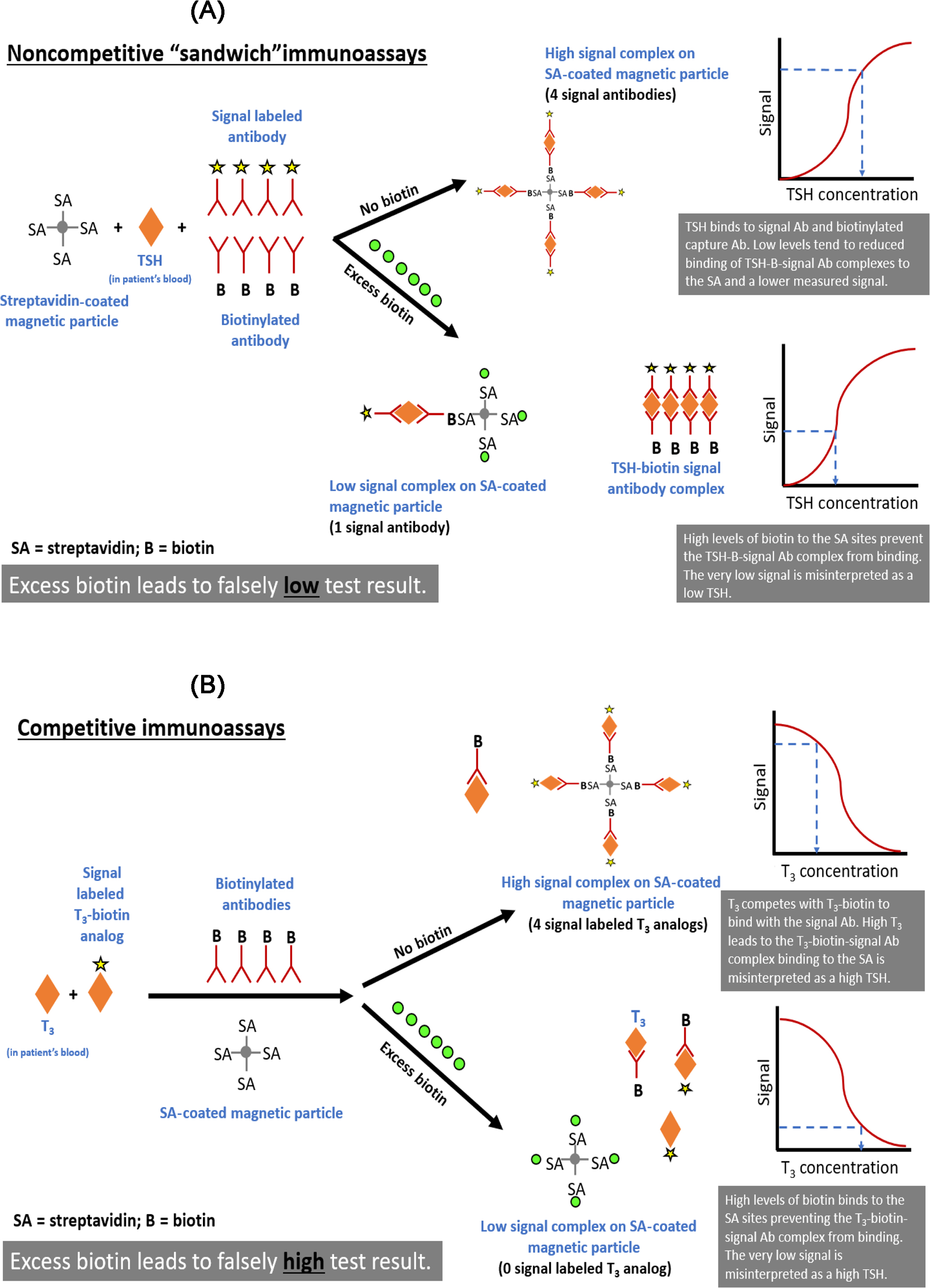
Biotin interference can affect assays for thyroid and reproductive hormones, immunosuppressants, and cardiac markers . Some immunoassays exhibit a higher threshold for biotin interference than is publicized, which could be due to differences in how biotin interference thresholds are calculated .
The lack of consistency between thyroid function test results and clinical symptoms is frequently attributed to biotin interference. Between 2016 and 2018, the Manufacturer and User Facility Device Experience (MAUDE) database maintained by the U.S. FDA reported that 78 of 92 incidents of potential biotin interference with immunoassays determined that biotin interference was “most likely” or “not ruled out” . However, it is difficult to verify biotin interference, and not all laboratories report incidents of potential biotin interference. Therefore incidents of biotin interference are probably underreported in the MAUDE database .
The concentration of biotin at which interference can occur varies from one assay to another . For example, biotin interferes with cardiac troponin assays at levels between 2.5 and 10,000 ng/mL . Most assays manufactured by Roche Diagnostics (Indianapolis, IN) are not significantly affected by biotin concentrations between 15.6 and 31.3 ng/mL. However, Roche thyroid and cardiac troponin T assays exhibit greater sensitivity to biotin interference . A high concentration of biotin and a biotin-sensitive assay are both required for a clinically relevant interference to occur. Although the Roche Diagnostics Elecsys Troponin T Gen 5 assay is sensitive to biotin interference , the risk of false-negative results is significantly lower than the misclassification risk due to the overall clinical performance of the assay .
Detection of biotin interference
All diagnostic algorithms should place test results within the context of the patient’s clinical condition. If test results are not consistent with the patient’s symptoms, it is possible that an analytical interference has occurred, and this possibility should be investigated. If biotin interference is suspected, biotin supplements should be discontinued, and the test should be repeated after biotin levels have normalized . Note that anomalies in immunoassay test results may be due to interferences other than biotin, and the steps that can be taken to identify the source of interference include:
- 1.
Perform a serial dilution study. Dilution decreases the biotin concentration and limits the likelihood that it will interfere with the assay. Serially dilutions should display the recovery of analyte consistent with each dilution. Nonlinear results suggest the presence of an interferent. Both the analyte and the interferent are diluted; the level of interference should be reduced at greater dilutions, and a reliable measure of the concentration occurs when the dilutions become linear with the signal. However, dilutions also affect the equilibrium between bound and free hormone fractions and can generate unreliable results.
- 2.
After exogenous biotin has cleared, the tests should be repeated, preferably using alternative assays/methods, such as direct assays for biotin concentrations by LC-MS or using immunoassays that are not based on biotin-streptavidin linkage.
- 3.
Various compounds can be added to the sample to adsorb biotin. For example, streptavidin agarose beads have been incorporated into the sample (10% by volume) before incubation for 60 min with intermittent mixing. After centrifugation, the supernatant was extracted, and the sample was retested. If was a discrepancy between the results before and after the depletion protocol, biotin interference was suspected. Studies have found that depletion protocols can be very effective . However, depletion protocols are considered lab-developed tests and should be thoroughly validated before use.
Additional approaches can be used to detect biotin interference, and no single approach is fully reliable because other interferents, such as heterophile antibodies, may also be present. In some laboratories, immunoassays that are not based on biotin-streptavidin linkage may be unavailable. Troponin assays are an example of time-critical laboratory results, and there may not be alternative methods available to help detect biotin interference.
The use of dietary biotin supplements is widespread. Laboratory staff and health care professionals should be fully informed of the risks of biotin interference and should implement appropriate procedures to minimize or eliminate this source of laboratory error. For example, the risk of an inaccurate diagnosis arising can be reduced by ensuring laboratories are operated in strict accordance with guidelines, patients are methodically assessed, and efficient and open communication channels are maintained between patients, clinicians, and laboratory staff. There is also a requirement for the implementation of continuous improvement opportunities and education that enhances laboratory testing protocols . Best practice documents should provide outlines for when patients should cease taking biotin-containing supplements prior to a blood test.
Anti-ruthenium and anti-streptavidin antibody interference
Electrochemiluminescent immunoassays that use ruthenium (Ru) chelate as labels can be a target of interfering antibodies (anti-Ru antibodies). The prevalence of anti-Ru antibodies interference has been estimated at <0.24% . The anti-Ru antibodies have been shown to cause falsely elevated free triiodothyronine (FT 3 ) concentrations on the Elecsys platform . Anti-Ru interference on free thyroxine (FT 4 ), thyroid stimulating hormone, and other hormones have also been described . Analysis of specimens with immunoassay methods that do not contain Ru label may be useful to detect anti-Ru antibodies .
For immunoassays that use streptavidin (SA) and biotin molecules, interference from antibodies against SA can cause erroneous hormone results . The prevalence of anti-SA antibody interference is unknown. Lam et al. reported discrepant thyroid hormone results that were determined to be due to IgM anti-SA antibodies. Anti-SA antibodies have been reported to cause spurious hyperthyroidism or misdiagnosis of Graves’ disease by interfering with anti-thyroid stimulating hormone receptor measurements . Using immunoassays without SA-biotin, dilution test, and polyethylene glycol precipitation procedures and incubation of the patient’s specimen with SA-linked agarose may be useful for the identification of anti-SA antibodies .
With anti-Ru and anti-SA antibody interference, sending specimens to the immunoassay manufacturer for further testing is beneficial to help identify these antibodies that can produce erroneous test results and subsequent patient misdiagnosis and mismanagement.
Serial monitoring and reference change value: Determining whether changes are significant
Laboratory tests for endocrine biomarkers frequently involve monitoring patients over time. It is essential to know whether significant changes in concentration of the analyte have occurred. Failure to recognize when changes are not significant can result in inappropriate changes in patient management. Conversely, failure to recognize significant changes in analyte concentrations can result in delayed diagnosis and treatment. The term reference change value (RCV) refers to the threshold for significance of changes in laboratory results .
Whether a change in biomarker concentration is significant can be calculated based on two factors: the physiological or intra-individual variation of the biomarker and the analytic variation of the test method. These two factors combined represent the total variability for each analyte or biomarker.
Physiological variation (intra-individual variation)
Intra-individual variation (CVi) has been calculated for many analytes; however, many common endocrine biomarkers are not well characterized with regard to their CVi values. Online resources that curate such lists are available; examples include westgard.com and biologicalvariation.eu . CVi values can vary from 0.5% for analytes such as plasma sodium, or as high as 37% for insulin .
Analytic variation
Analytic variation (CVa) varies considerably between analytes, methodologies, instruments, and laboratories. The CVa for any test performed in a clinical laboratory is known and typically can be found in the manufacturer’s product literature or the method standard operating procedure. Variability may change across the dynamic range of the assay, so the precision at high concentrations may not be the same as at low concentration. For some biomarkers, CVa values are near 1%. However, for biomarkers that have very low concentrations, the CVa can be as high as 20%. When the CVa of an assay exceeds 20%, the method is generally considered only semiquantitative.
Calculating significant variation
The calculations presented here assume that the same methodology and instrument are being used to monitor a patient. Comparing serial results from different laboratories, methodologies, or instruments is difficult and should be avoided. The total variation is a product of the intra-individual and analytic variation, but unfortunately, these two parameters cannot be added together directly. CV values (or standard deviations, SD) must be converted to variances to be additive . Fortunately, the conversion of CV to variance is only a matter of summing the squares of the two CV values, then taking the square root:
First, calculate the total variation:
For two results to be significantly different (at P <.05), they have to differ by at least 2.8 SD. Multiply the CV%×2.8. This is the percentage difference between the two results required to be statistically different.
The difference between the two results is calculated as (Result 1−Result 2)/Result 1×100%.
Some examples of when this approach is helpful are outlined below. Each example is based on a 95% confidence value threshold ( P <.05).
Example 1—Is the statin working?
A patient taking atorvastatin has a steady-state total cholesterol of 275 mg/dL. After changing to fluvastatin, the total cholesterol lowered to 235 mg/dL. The ordering physician is satisfied as a result is now below the 239 mg/dL threshold recommendation for “intermediate risk.” Is this change significant?
For total cholesterol, the intra-individual variation is 5.9%, and the analytic variation is 2.4%.
The least significant change will be 6.4%. Taking 6.4% and multiplying by 2.8 yields 17.9%.
The minimum change needed for significance is 17.9%.
For this patient, the results are 275 and 235 mg/dL.
The percent difference is calculated to be: (275–235)/275×100%=14.5%.
Because the change was only 14.5% and a minimum 17.9% change is needed for significance, this reduction in total cholesterol can be attributed to a combination of physiological and analytic variability.
Example 2—Is the DPP-4 inhibitor working?
A physician has a diabetic patient with a hemoglobin A1c of 9.1% after being on a sulfonylurea. While watching sports on television, a physician saw an advertisement for sitagliptin, which claimed that adding this medicine could reduce A1c by 0.7 percentage points.
If the median intra-individual CVi is 1.3% and the analytic CVa is 3.8%, would the reduction in A1c from the advertisement be considered significant for an individual patient?
The least significant change will be 4.0%. Taking 4.0 and multiplying by 2.8 yields 11.2%.
The minimum change needed for significance is 11.2%.
For this patient with a result of 9.1% A1c, a 0.7 percentage point reduction would be 8.4%.
The percent difference is calculated to be: (9.1–8.4)/9.1×100%=7.7%
Therefore a change in A1c of 0.7 percentage units would be only 7.7%, whereas a result would need to be 11.2% different to be significant for an individual patient.
Example 3—Sestamibi scan or surgical consult?
A patient has a PTH concentration of 95 pg/mL, which is above the laboratory reference interval of 10–65 pg/mL. Six months later the PTH is measured again, this time at 156 ng/mL. Is this change significant?
For PTH, the intra-individual variation is 25% and the analytic variation is 12%.
The least significant change will be 27.7%. Taking 27.7 and multiplying by 2.8 yields 78%.
The minimum change needed for significance is 78%.
For this patient the results are 95 and 156 mg/dL.
The percent difference is calculated to be: (95–156)/95×100%=−65%.
Because the change was only 65% and a minimum 78% change is needed for significance, this increase in PTH could be due to physiological and analytic variability.
Automated calculations
The RCV tool is not particularly computationally challenging. Performing the operation manually to determine 95% confidence is simple and determining the percent likelihood of a significant change is slightly more challenging but within the capability of any modern computing device. This calculation can be implemented in laboratory information systems (LIS), physician electronic health record systems, Excel spreadsheets, and even smartphone apps .
Postanalytical errors
Postanalytical errors occur after a laboratory result has been generated by an analytical instrument. Incorrect transcription or data entry of laboratory results is a major source of postanalytical errors. Studies of point-of-care laboratory tests, where results are often manually entered into the patient record, have revealed error rates of approximately 5 in 1000 recorded results . Although transcription errors remain a problem in point-of-care settings where the tests are visually interpreted (e.g., fecal occult blood, pregnancy tests, urine dipsticks), most core clinical laboratories use automated instrument platforms that are digitally interfaced to the LIS, which, in turn, is interfaced with the electronic medical record (EMR), so data are passed from the analytical instrument to the patient record without human intervention. Digital interfaces greatly reduce the frequency of errors in the postanalytical data processing. Many point-of-care instruments are capable of digital communication with the LIS or EMR, as well. These technological advances have vastly improved the integrity of the postanalytical processing of laboratory results compared to manual entry.
Preanalytical and analytical errors can affect the accuracy of a laboratory result; however, most of these errors are identifiable, and their frequency can be minimized by vigilance and engineering. On the other hand, many postanalytical errors are very difficult to detect and prevent because they involve how a laboratory result is interpreted and used to influence the treatment of a patient. For most laboratory tests, the involvement of laboratory personnel (e.g., technologists, directors, pathologists) ends when the test result is reported. Surveys have shown that consultation with laboratory personnel or a pathologist is not common even when the requesting physician is uncertain about which test to order or the meaning of a test result . To make matters worse, medical students in the United States receive an average of just 9 h of instruction in the proper selection and interpretation of laboratory tests . Errors in the interpretation of laboratory test results fall into three broad categories:
- 1.
Misunderstanding of the reference range, and the various ways “expected,” or “desired” results are presented with a laboratory result;
- 2.
Failure to correctly interpret the statistical probability that a laboratory result correctly classifies the disease status of a patient, which can lead to incorrect assessment of risk and, often, unnecessary follow-up testing and procedures;
- 3.
Misinterpretation of the meaning of an abnormal laboratory result, and the specific diagnostic possibilities it might reveal.
The third category is far beyond the scope of this introductory chapter, as it requires detailed knowledge of endocrine function and testing, which will be addressed in subsequent chapters. However, the first two categories are more general and merit introductory review because they apply to virtually all laboratory tests.
Reference ranges, normal values, and desired results
Accreditation agencies and the Centers for Medicare and Medicaid Services require all laboratory test results to be presented with a reference range or other interpretive guideline to provide advice about whether the patient’s result is normal or abnormal. However, the terminology in reference ranges or interpretive guidelines can be confusing, particularly to laypersons. For example, a “positive” laboratory test is typically an abnormal result, whereas in common (nonmedical) usage “positive” usually means something good. The same is true for “negative,” which is good news in a medical context, but in common usage denotes something undesirable. The use of “normal” is imprecise because it implies “healthy,” and laboratory results that fall within “normal limits” do not necessarily indicate health, just as results that exceed those limits can occur in healthy patients. Even the term “reference range” is slightly problematic, since it raises the question: “in reference to what?” The answer to that question can differ from one laboratory test to another.
Fundamentally, reference ranges reflect interpretative advice that is based on one or more of the following:
- 1.
Population-based reference intervals that are generated by testing healthy individuals in a population that mirrors the characteristics of the patients to which the reference interval is being applied; or
- 2.
Desired results based on research and epidemiological data that indicate what level of a particular biomarker is associated with good health or absence of disease.
Population-based reference intervals typically reflect the central 95% of results obtained by testing a suitable reference population. The selection of an appropriate reference population may be the most critical challenge in generating a reference interval because it should account for changes in analyte concentration that may occur due to age, sex, ethnicity, geography, diet, or other factors. Fortunately, differences based on these factors are minimal or nonexistent for most common laboratory tests. However, some analytes show marked differences based on age, sex, and sometimes ethnicity. Influences due to geography, diet, and other factors are relatively rare but do exist. For example, vitamin D 3 is produced in the skin by the action of UV light on 7-dehydrocholesterol, so people who live at latitudes that receive more direct sunlight typically have higher vitamin D levels . People who live at high altitudes usually compensate for the lower oxygen tension by hematopoiesis and have higher average hemoglobin . Vegetarian diets have been shown to lower average total, low density lipoprotein, and high density lipoprotein cholesterol levels .
A common method used to generate a reference range is to calculate the mean and SD of results in the reference population and then set the reference range as the mean±2 SD. Note that defining the central 95% as the mean±2 SD is valid only if the data conform to a Gaussian distribution. While some analytes may fit a bell-shaped distribution curve, the data are usually unevenly distributed (i.e., skewed toward higher or lower values). For this reason, the CLSI recommends, in their 2010 document EP28-A3c “Defining, Establishing, and Verifying Reference Intervals in the Clinical Laboratory; Approved Guideline-Third Edition,” that reference intervals be determined by a nonparametric method that involves discarding the highest and lowest 2.5% of values, with the remaining limits defining the central 95% reference interval. The drawback of this approach is that it requires at least 120 reference values. It may be difficult for most laboratories to recruit enough volunteers to donate specimens for a reference range study. The mean and SD of Gaussian distribution can be estimated with approximately 30 data points, but if the distribution of results is not truly Gaussian, then the mean±2 SD approach will generate a misleading reference interval.
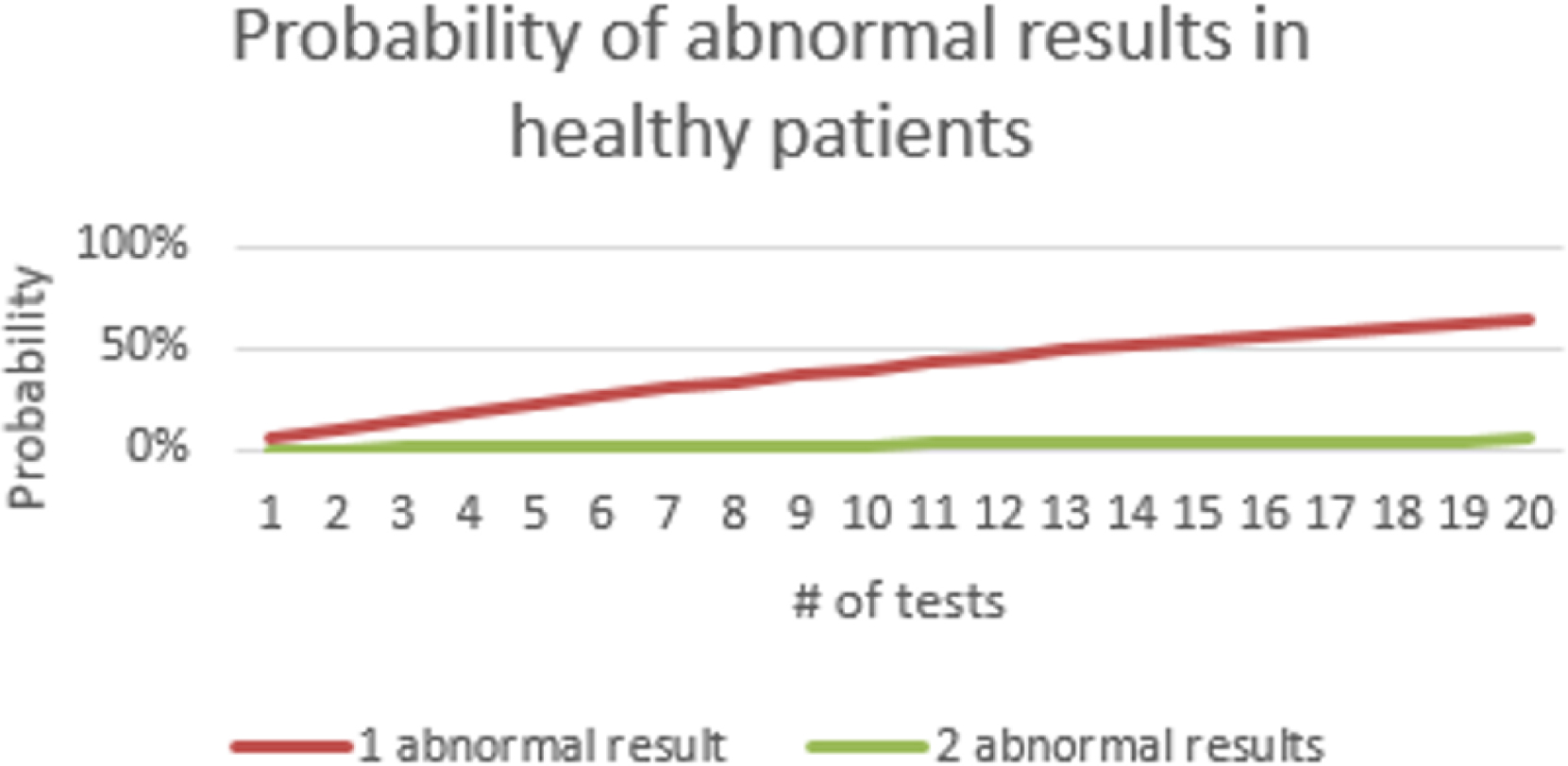
There is another subtle issue in the way that “central 95%” reference intervals are generated: the range does not include all healthy subjects. On the contrary, 5% of the reference population had test results that exceeded the newly established reference interval. Therefore their test results would be considered abnormal. From a statistical perspective, the odds that a perfectly healthy patient will have an abnormal result in a single laboratory test is about 1 in 20. If two tests are performed on a healthy patient, the odds go up to about 1 in 10 that at least one of the results will be abnormal. For a basic metabolic profile, which includes eight tests, the statistical probability that at least one result in a healthy patient will be outside its reference range is 0.33 (33%). In a comprehensive metabolic profile, which includes 14 tests, it is more likely than not ( probability =0.51) that a healthy patient will have at least one test result that is outside the limits of its reference range. The relationship between the number of tests and the probability of an abnormal result is illustrated in Fig. 1.4 .