Maturity-Onset Diabetes of the Young
Andrew T. Hattersley
The early clinical descriptions recognized that maturity-onset diabetes of the young (MODY) was a monogenic form of diabetes with a single gene mutation causing diabetes within a family. Our understanding of MODY has been transformed by defining the genes involved in this condition. MODY initially was defined solely by clinical criteria, but now a genetic subclassification is possible based on the description of six genes in which mutations have been described. With the description of the etiologic genes, there have been new clinical insights, and with each gene resulting in the description of discrete clinical and physiologic entities for mutation. The definition of the underlying molecular genetics frequently helps determine the likely clinical course, prognosis, and best treatment options. Recent classifications of diabetes by the American Diabetes
Association (ADA) and the World Health Organization (WHO) (1) have recognized this by classifying MODY now as discrete subtypes of diabetes arising from mutations in specific β-cell genes. MODY is probably the first area of diabetes in which molecular genetics has played a clear clinical as well as research role.
Association (ADA) and the World Health Organization (WHO) (1) have recognized this by classifying MODY now as discrete subtypes of diabetes arising from mutations in specific β-cell genes. MODY is probably the first area of diabetes in which molecular genetics has played a clear clinical as well as research role.
EARLY DESCRIPTIONS OF MATURITY-ONSET DIABETES OF THE YOUNG
In the pre-insulin era, it was recognized that some patients with diabetes did not die and that these patients typically had a parent with diabetes. Cammidge noted in 1928 that “a dominant inheritance is almost invariably associated with a good prognosis” (2). It is likely that this is the first recognition of a clinical subgroup that was later known as MODY.
Strongly inherited young-onset familial diabetes subsequently was recognized and studied by Fajans in the 1960s and 1970s (3,4). In 1974, Tattersall recognized that an autosomal dominant inheritance was a feature of this condition (5). The term MODY was coined in 1975, although initially this stood for maturity-onset type diabetes of young people (6). Subsequently, a large number of families with variable phenotypes were described throughout the world. Although some of the phenotypic variation resulted in the use of different definitions, it became apparent that there was clinical heterogeneity in MODY, even if only white families with an age of onset of 25 years were considered (4,7). This clinical heterogeneity is now explicable in terms of genetic heterogeneity (8).
CLINICAL DEFINITION OF MATURITY-ONSET DIABETES OF THE YOUNG
In the vast majority of patients, it is now possible to define MODY as diabetes associated with β-cell dysfunction resulting from a specific mutation in a MODY-related gene. However, it is clearly not feasible to perform genetic studies on all subjects, and hence clinical criteria still are necessary to identify those patients in whom a mutation is likely. In addition, some subjects have MODY but the etiologic genes have not yet been defined. Criteria we propose are those used in genetic studies in the United Kingdom and France (8).
Early Onset Non-Insulin-Dependent Diabetes
By definition, a family is considered to have early onset diabetes if at least one and ideally two members of the family are diagnosed with diabetes before the age of 25 years, although it is recognized that other family members, particularly in older generations, may be diagnosed later.
Patients are considered non-insulin-dependent if 5 years after diagnosis they either are not receiving insulin treatment or are insulin treated but have significant levels of circulating C-peptide. Family members are considered to be affected either if they had diabetes by WHO criteria or, particularly in the case of glucokinase mutations, if they had fasting hyperglycemia (>6 mmol/L or >108 mg/dL) (9,10).
Autosomal Dominant Inheritance
The minimal criterion for autosomal dominant inheritance is the occurrence of diabetes in two generations, although most MODY families have at least three or more generations affected. In an autosomal dominant condition, only one of the two parents of an affected child should be affected. Therefore, considerable caution should be taken before defining a family in which both parents have diabetes as a MODY family, especially because children who inherit a “double gene dose” of type 2 diabetes from both parents also may have an earlier onset of diabetes (11).
Other Features
While early-onset, non-insulin-dependent diabetes and an autosomal dominant inheritance have typically been used for the definition of MODY, other features can help in identifying whether a particular family has MODY. The most important features follow:
β-cell dysfunction: β-cell dysfunction rather than insulin resistance is a characteristic of MODY. A young subject with acanthosis nigricans, the cutaneous marker of marked insulin resistance, is very unlikely to have MODY.
Lean-body habitus: Subjects with MODY mutations do not need to be obese to develop diabetes, in marked contrast to most subjects with childhood or early onset type 2 diabetes (12). However, MODY cannot be excluded if a subject is obese, as most series suggest that obesity is similar to that seen in the normal population. However, it is unlikely to be a consistent feature of diabetes throughout a family.
DEFINITION OF THE GENETIC BASIS OF MATURITY-ONSET DIABETES OF THE YOUNG
The development of modern genetic techniques centered on the use of the polymerase chain reaction in the early 1990s has considerably helped in defining the genes in MODY. The definition of genes in monogenic MODY families was always likely to be considerably easier than defining genes in polygenic type 1 and type 2 diabetes, in which there are multiple genes and a large environmental component. The early onset favored the collection of large, multigeneration pedigrees that greatly facilitated analysis and gave a single family sufficient power to define a new locus or gene (9,13,14). Candidate genes were also relatively easy to define, as all families had a β-cell defect.
The breakthroughs in defining the genetic etiology came through the use of linkage methods. A candidate-gene approach was used in defining the role of the glucokinase gene in French and English MODY pedigrees (9,10). The major breakthrough, however, was the use of linkage to localize etiologic genes with a positional mapping approach that identified susceptibility loci on chromosome 20q (MODY1) and 12q (MODY3) (13,15). The MODY gene on 12q was defined as being hepatic nuclear factor (HNF) 1α (16). This was a seminal finding that led to the rapid recognition that HNF4α was the MODY1 gene (17). It also clearly indicated that HNF1β (MODY5), which forms heterodimers with HNF1α, was also an excellent candidate gene. The role of the HNF in β-cell function had previously been unsuspected. The description of insulin-promoter factor (IPF) 1 (also known as PDX1) as the MODY4 gene arose from observations in a family initially investigated because the proband had pancreatic agenesis, a condition seen in the IPF1 knockout mouse (14,18). It was shown that heterogeneity for a severe mutation caused MODY, while homozygosity for this mutation caused pancreatic agenesis.
A variety of approaches have therefore led to the identification of the genes involved in MODY. These observations opened new areas of understanding and research on β-cell physiology, pathophysiology, and genetics.
RELATIVE PREVALENCE OF THE DIFFERENT SUBGROUPS OF MATURITY-ONSET DIABETES OF THE YOUNG
Large national collections, particularly in the United Kingdom and France, allow assessment of the relative prevalence of the different subgroups of MODY. Figure 26.1 shows the relative contribution of the six known genes in United Kingdom pedigrees that have been collected principally from adult hospital clinics (19).
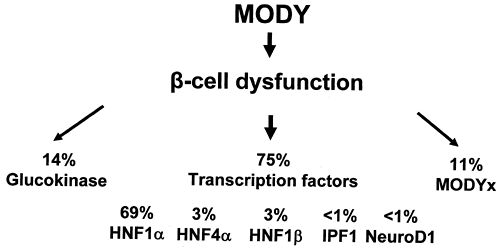 Figure 26.1. Distribution of MODY in the United Kingdom MODY collection. (Data from 131 families from Frayling TM, Evans JC, Bulman MP, et al. Beta-cell genes and diabetes: molecular and clinical characterization of mutations in transcription factors. Diabetes 2001;50[Suppl 1]:S94–S100; and A. T. Hattersley and S. Ellard, unpublished.) |
Although the prevalence of the mutations varies, HNF1α (MODY3) is the commonest cause in most MODY series. This depends on the strictness of the criteria for MODY used when recruiting families, the predominant method of recruiting families, and racial origin. In two large collections from France and Italy, glucokinase (MODY2) is the most prevalent subtype (20,21). This probably represents the detection of asymptomatic, mild fasting hyperglycemia in children based on recruitment from pediatric clinics or by screening young children in family studies (22). In contrast, the majority of other MODY series are recruited from adult diabetes clinics, where the majority of patients have symptomatic diabetes, so MODY3, with more marked hyperglycemia, is more common. It does appear that, even allowing for differences in diagnostic criteria and ascertainment, the prevalence of the known MODY genes is considerably lower in Japanese and Chinese series than in series of European whites.
As the different genetic subgroups of MODY have clear differences in clinical and physiologic phenotypes, they are discussed separately below and summarized in Table 26.1. A genetic classification is clinically useful, as the phenotypic differences between MODY2 and MODY3 are as marked as the differences between type 2 diabetes and either MODY2 or MODY3.
TABLE 26.1. Comparison of the Different Subtypes of Maturity-Onset Diabetes of the Young | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
HNF4α MUTATIONS (MATURITY-ONSET DIABETES OF THE YOUNG-1)
Molecular Genetics
MODY1 was the first genetic locus to be defined in MODY (13). The localization of the gene to the long arm of chromosome 20 by Bell et al. (13) was possible because of the collection of DNA samples and careful longitudinal study of the extensive RW pedigree by Fajans (23). Fine mapping of this gene was difficult, as linkage to MODY1 was rare (10). The major breakthrough in defining the MODY1 gene was the identification of the MODY3 gene as HNF1α (16). This led to the rapid recognition that HNF4α was the MODY1 gene and to the identification of a nonsense mutation Q268X that co-segregated with MODY in the RX pedigree (17) (see below).
Mutations in HNF4α are considerably less common than mutations in HNF1α, with fewer than 20 mutations being described during the first 5 years following the description of the gene [reviewed in reference (24)]. These mutations are distributed throughout the gene and consist of nonsense, frameshift, and missense mutations. Recently, it has been shown that the main HNF4α isomer in the β-cell uses a far-upstream promoter and alternative exon 1 (25). This P2 promoter has HNF1 and IPF1 binding sites. The critical regulatory role of these binding sites in the β-cell has been established by studies in HNF1α knockout animals and the finding of a mutation that causes MODY in the IPF1 binding site (25,26).
Phenotype of Patients with HNF4α Mutations
CLINICAL CHARACTERISTICS
The clinical phenotype of HNF4α mutations is far more similar to the phenotype seen with HNF1α mutations than the phenotype seen with the glucokinase mutations (Table 26.1). Children younger than 10 years of age who have HNF4α mutations usually have normal glucose tolerance and develop diabetes in adolescence or early adulthood. In some families, the age at diagnosis is older than that in HNF1α families, but this is not a consistent finding (27). Patients show worsening glycemia with increasing age as the result of progressive β-cell dysfunction.
HNF4α is expressed in a wide range of cell types and tissues. Secretion of both glucagon from the α-cells and the pancreatic polypeptide (PP) cells is also reduced in patients with MODY1 (28).
PENETRANCE OF MUTATIONS
HNF4α generally has a high penetrance, with the majority of subjects with a diagnosis of diabetes by the age of 25 years. Teenagers and young adults who have carried the mutation but who had not developed diabetes on repeated glucose tolerance testing have been described. In some families, the older age of diagnosis probably reflects reduced penetrance of specific mutations (27,29).
TREATMENT
As in patients with HNF1α mutations, those with HNF4α mutations show a progressive deterioration in glycemia and frequently require oral agents and insulin. Some patients have been successfully maintained for decades on sulfonylureas (30) and, like patients with MODY3, may be sensitive to the hypoglycemic effects of sulfonylurea therapy.
COMPLICATIONS
Both microvascular and macrovascular complications are frequent. The frequency is thought to be similar to that in subjects with type 1 or type 2 diabetes (4).
EXTRAPANCREATIC FEATURES
The main extrapancreatic manifestations described result from reduced transcription of hepatic target genes of HNF4α. Reduced concentrations of the apolipoproteins apoAII, apoCIII, and possibly apoB are found in patients with MODY1 but not in patients with type 2 diabetes (31,32). Triglyceride levels are decreased in patients with MODY1 in contrast to patients with type 2 diabetes, in whom levels are usually increased. This elevation probably reflects decreased lipoprotein lipase activity resulting from the reduction in apolipoproteins (31). In contrast to patients with mutations in HNF1α, low renal threshold for glucose has not been reported in patients with MODY1.
PATHOPHYSIOLOGY
Patients with HNF4α mutations have progressive β-cell dysfunction. This β-cell dysfunction is characterized by an inability to increase insulin secretion when blood glucose levels are high (33). In contrast to HNF1α, prolonged hyperglycemia (18 hours) does not prime the β-cell in nondiabetic individuals (33).
CLASSIFICATION
The separation of patients with HNF4α mutations from patients with mutations in other transcription factors would normally require molecular genetic diagnosis. The ADA classification proposes that an etiologic diagnosis be used, for example, genetic defect in β-cell function; chromosome 20 HNF4α.
PREVALENCE
Mutations in HNF4α are far less common than mutations in HNF1α. In the United Kingdom, families with HNF4α mutations represent 2% to 4% of MODY families (Fig. 26.1).
Disease Mechanisms in Maturity-Onset Diabetes of the Young-1
FUNCTIONAL CHARACTERISTICS OF MATURITY-ONSET DIABETES OF THE YOUNG-1 MUTATIONS
The functional characteristics of HNF4α mutations associated with MODY1 have been studied extensively [reviewed in reference (24)]. The principal mutation mechanism is haploinsufficency, with the majority of mutations showing a considerable reduction of transcription activity in vitro: They are not “dominant-negative” mutations, because they do not significantly interfere with the activity of wild-type HNF4α. The HNF4α mutation associated with later-onset type 2 diabetes shows intermediate activity in vitro between wild-type and the mutations causing MODY (27).
Mechanisms Involved in Development of Diabetes in Maturity-Onset Diabetes of the Young
The mechanisms resulting in heterozygous mutations in the HNF4α gene that cause β-cell dysfunction are not fully established. Studies by Stoffel and Duncan (34) using embryonic stem cells have established that HNF4α regulates the expression of genes encoding components of the glucose-dependent insulin-secretion pathway, including glucose transporter 2; the glycolytic enzymes aldolase B and glyceraldehyde 3-phosphate dehydrogenase; and liver pyruvate kinase. The progressive loss
of β-cell function is hard to explain purely by altered levels of glucose transport and metabolism and may reflect a progressive reduction in β-cell mass due to altered β-cell turnover.
of β-cell function is hard to explain purely by altered levels of glucose transport and metabolism and may reflect a progressive reduction in β-cell mass due to altered β-cell turnover.
GLUCOKINASE MUTATIONS (MATURITY-ONSET DIABETES OF THE YOUNG-2)
Molecular Genetics
GLUCOKINASE AS A CANDIDATE GENE FOR MATURITY-ONSET DIABETES OF THE YOUNG
Glucokinase was the first MODY gene identified. The role of glucokinase in insulin secretion made it an ideal candidate gene for MODY. Glucokinase is one of four isoforms of hexokinase, enzymes that catalyze the first step in the metabolism of glucose: its phosphorylation to glucose-6-phosphate. Glucokinase is expressed principally in pancreatic β-cells and in hepatocytes. In β-cells, glucose phosphorylation is closely linked with the initiation of insulin secretion (35,36). Unlike the other hexokinases, glucokinase has a low affinity for glucose (K m of 5.5 mmol/L or 99 mg/dL plasma glucose concentration) and is not inhibited by its product. These features allow β-cells and hepatocytes to change rates of glucose phosphorylation at physiologic glucose concentrations (4–15 mmol/L or 72–270 mg/dL). Glucokinase had consequently been termed the pancreatic glucose sensor (36).
MUTATIONS OF THE GLUCOKINASE GENE CAUSE MATURITY-ONSET DIABETES OF THE YOUNG
The characteristic of glucokinase strongly suggested that this might be an important candidate gene in type 2 diabetes. Tanizawa and colleagues in Permutt’s laboratory at Washington University in St. Louis cloned the human gene and showed that it had a structure similar to that of the rat gene, with 12 exons and discrete pancreatic and hepatic promoters (37). The identification of two microsatellite polymorphisms flanking the glucokinase gene led to the description in early 1992 of linkage in French and English MODY pedigrees (9,10).
MOLECULAR GENETICS OF THE GLUCOKINASE GENE
The demonstration of linkage between MODY and the glucokinase gene was quickly followed by the characterization of the human gene and the detection of mutations (38,39,40). The first mutation was reported in 1992 (38), and more than 100 mutations of the glucokinase gene have now been described in many populations, with the majority found in France and Italy (20,21,38,39,40,41,42,43). All these mutations have been associated with mild to moderate hyperglycemia.
It is interesting that one mutation is associated with the opposite phenotype, that is, hypoglycemia associated with hyperinsulinemia (44). This is due to a gain-of-function mutation that results in overproduction of insulin. Homozygous loss-of-function mutations in the glucokinase gene have recently been shown to result in permanent neonatal diabetes that is severe and requires insulin treatment from birth (45).
Mutations associated with hyperglycemia are distributed throughout the gene, and different mutation mechanisms, with nonsense, missense, frameshift, and splice-site mutations, having all been described. The mutations associated with MODY2 have been shown to have reduced activity when expressed in vitro. Many mutations alter the maximal activity of the enzyme for glucose phosphorylation or alter its affinity for glucose (46). Other mutations, distant from the glucose binding cleft of the enzyme (encoded by exons 5, 6, 7, and 8), either can alter the structure by a major alteration in protein structure as a result of nonconservative changes in amino acid or can alter adenosine triphosphate phosphorylation (47,48).
Phenotype of Patients with Glucokinase Mutations
CLINICAL CHARACTERISTICS
Despite the wide variety of mutations (49), alterations in the glucokinase gene result in a discrete phenotype, summarized in Table 26.1. An increased fasting blood glucose is a consistent feature, and the majority of patients have blood glucose values within a tight range of 6 to 8 mmol/L or 108 to 144 mg/dL (Fig. 26.2). A fasting glucose level below 5.5 mmol/L is extremely rare in MODY2 patients, and no patients with mutations have had fasting glucose values consistently below 5.5 mmol/L (50). There is a mild deterioration in fasting blood glucose levels with age, but blood glucose levels in patients in their eighth and ninth decades still rarely exceeded 9 mmol/L (50,51,52). This mild hyperglycemia is present very early in life, and patients have been described who have hyperglycemia in the first weeks of life (53). Recent observations showing altered birth weight and fetal growth support the concept that these changes in the β-cell function are present before birth (43).
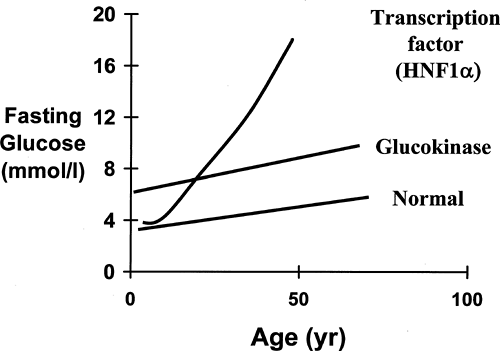 Figure 26.2. Diagrammatic representation of the change in fasting glucose with age in untreated patients with HNF1α and glucokinase mutations. (Based on data from Hattersley AT. Maturity-onset diabetes of the young: clinical heterogeneity explained by genetic heterogeneity. Diabet Med 1998;15:15-24; Stride A, Vaxillaire M, Tuomi T, et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia 2002;45:427–435; and Pearson ER, Velho G, Clark P, et al. Beta-cell genes and diabetes: quantitative and qualitative differences in the pathophysiology of hepatic nuclear factor-1alpha and glucokinase mutations. Diabetes 2001;50[Suppl 1]:S101–S107.) |
Patients with glucokinase mutations are usually asymptomatic throughout their life and rarely have symptoms of hyperglycemia. The vast majority will be detected by screening either for routine medical examinations, during pregnancy, or family screening when MODY is suspected (9,54). In most patients, the age of diagnosis is the age at which they are first tested. While being diagnosed young is supportive of a glucokinase mutation, the fact that a patient is diagnosed only in old age should not be used to exclude the diagnosis. More than in any other type of diabetes, MODY2 may be diagnosed decades after the age of onset.
TREATMENT
Patients with glucokinase mutations rarely need any pharmacologic treatment (51,52). The majority of patients with glucokinase mutations (>85%) are treated with diet alone. Avoiding unrefined carbohydrate may not be as important as in patients with type 2 diabetes, because most patients with glucokinase mutations show little increment in their blood glucose on receiving 75 g of glucose in an oral glucose tolerance test (50,52,55). Another interesting observation is that body mass index makes little difference to the level of glycemia in patients with glucokinase mutations (50,51). This is in marked contrast to patients with type 2 diabetes, in whom weight loss can very effectively reduce glycemia. In patients with glucokinase mutations, this is probably because the fasting hyperglycemia is due to a defect in glucose sensing; thus, as in the healthy subject, increased insulin resistance as a result of obesity results in compensatory hyperinsulinemia rather than hyperglycemia. The only time that patients with glucokinase mutations are likely to consistently receive treatment is during pregnancy (see below).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


