Major Platelet Glycoproteins: Platelet Glycoprotein Ib-IX-V
Michael C. Berndt
Robert K. Andrews
In the past 5 years, understanding of the structure and function of the platelet glycoprotein (GP) Ib-IX-V complex1,2 has advanced in a number of key areas that should facilitate future therapeutic targeting in thrombotic and other diseases. This chapter focuses on new experimental and clinical studies involving mechanisms regulating GPIb-IX-V expression, ligand binding and signaling, the role of GPIb-IX-V in hemostasis, and new nonhemostatic functions for this receptor. In hemostasis and thrombosis, engagement of GPIbα (the ligand-binding subunit of GPIb-IX-V) by von Willebrand factor (vWF) leads to platelet activation, secretion of platelet agonists such as ADP and thromboxane A2, and initiation of intracellular signaling pathways resulting in activation of platelet integrins, mainly αIIbβ3 (GPIIb-IIIa), the receptor that binds fibrinogen or vWF and mediates platelet aggregation (thrombus formation) (see Chapter 15B). Critical to the structure and function of GPIb-IX-V are specific adaptations enabling interaction between GPIbα and vWF or other ligands at high physiologic or pathologic shear rates in the bloodstream. This underlies much of the current interest in GPIb-IX-V as a target for antiplatelet therapy in heart attack and stroke.
GPIb-IX-V STRUCTURE
The GPIb-IX-V complex is made up of the ligand-binding GPIbα subunit, disulfide bonded to GPIbβ and noncovalently associated with GPIX and GPV.1,2 Key structural domains are illustrated in FIGURE 26A.1. The stoichiometry appears to involve each GPIbα linked to 1 or 2 GPIbβ subunits via adjacent Cys residues in the extracellular domain of GPIbα nearby the outer face of the plasma membrane.2,3 There are equivalent levels of GPIX and GPIb expression and approximately half as many copies of GPV;4 GPIbα, GPIbβ, and GPIX are required for stable surface expression.2
GPIb-IX-V EXPRESSION AND SHEDDING
Expression of GPIb-IX-V is regulated by ectodomain shedding (FIGURE 26A.1). ADAM17 is one of the sheddases involved in release of the GPIbα ectodomain (glycocalicin).5,6 GPIbα is constitutively shed from circulating platelets, and glycocalicin levels in plasma are 1 to 3 µg/mL. Shedding is increased upon platelet activation or aging in vivo or ex vivo.7 Thrombin, ADAM10, or ADAM17 cleave the ectodomain of GPV within the same region.5,8 Deficiency of GPV in mice enhances the capacity of thrombin to stimulate platelets via GPIbα.9
GPIb-IX-V: EXTRACELLULAR BINDING PARTNERS
The primary GPIb-IX-V ligand is vWF, a multivalent multimeric adhesive protein secreted from platelets and endothelial cells. Each mature subunit of vWF (˜275 kDa) is composed of conserved domains (in order, D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2). The vWF-A1 domain, encompassing an intramolecular Cys509-Cys595 disulfide bond, contains the binding site for GPIbα. The vWF-A3 domain binding contains a binding site for collagen and the vWF-C1 domain contains an Arg-Gly-Asp (RGD) sequence that mediates binding to the activated integrin, αIIbβ3. Binding of vWF-A1 to GPIb-IX-V has been investigated using proteolytic fragments of native vWF or GPIbα,10,11 crystal structures of GPIbα/vWF-A1 interactive domain complexes,12,13 mutagenesis studies examining this interaction under shear conditions,14 and in silico modelling of individual receptor/ligand bonds.15,16 Together, these studies have provided detailed insight into how vWF can support platelet adhesion under shear conditions, by conformational shear-dependent activation of catch-slip bonds,16 which enhance or decrease individual bond strength to regulate platelet attachment or detachment, respectively. It has also been demonstrated by structural studies how small molecule inhibitors that bind GPIbα within the N-terminal leucine-rich repeat domain can allosterically block vWF binding.17
Other adhesive ligands for GPIb-IX-V are the counterreceptors P-selectin,18 expressed on activated platelets and activated endothelial cells, and the integrin αMβ2 (Mac-1)19 expressed on leukocytes. Binding of P-selectin involves the anionic sulfated tyrosine sequence of GPIbα, analogous to an N-terminal sulfated sequence within the counterreceptor for P-selectin on leukocytes, P-selectin glycoprotein ligand-1 (PSGL-1). The αM I-domain (homologous to vWF-A domains) contains the binding site for GPIbα.20 This interaction is involved in recruitment of leukocytes to adherent platelets at sites of injury or infection (P-selectin on activated platelets binding to PSGL-1 on leukocytes leads to activation of αMβ2) and could also be involved in platelet clearance. In this regard, clustered carbohydrate structures of GPIbα on platelets refrigerated ex vivo are recognized by αMβ2 on hepatic macrophages, and provides at least one mechanism for the cold storage lesion that results in rapid clearance of transfused cold-stored platelets.21
There is also evidence that collagen can bind GPV,22 although the physiologic importance of this interaction is not yet established. GPV deficiency in mice does not result in a bleeding phenotype23 and there is at most a limited effect on hemostatic
function.24 Nevertheless, release of GPV by metalloproteinasemediated ectodomain shedding in activated platelets or by thrombin (FIGURE 26A.1) has led to investigation of plasma GPV as a platelet-specific marker of platelet activation or coagulation in vivo.25
function.24 Nevertheless, release of GPV by metalloproteinasemediated ectodomain shedding in activated platelets or by thrombin (FIGURE 26A.1) has led to investigation of plasma GPV as a platelet-specific marker of platelet activation or coagulation in vivo.25
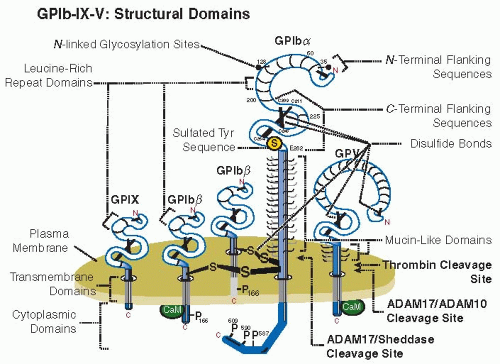 FIGURE 26A.1 GPIb-IX-V: Structural domains. Illustration of the GPIb-IX-V complex, consisting of GPIbα, GPIbβ, GPIX, and GPV subunits, showing leucine-rich repeats, disulfide-looped N– and C-terminal capping sequences, transmembrane domains, and cytoplasmic domains. GPIbα contains a mucin-like domain elevating the major ligand-binding domain (FIGURE 26A.2). The anionic sulfated tyrosine sequence (residues 269 to 282) contains three sulfated tyrosine residues (Tyr275, Tyr276 and Tyr278). ADAM17 or other sheddases cleave N-terminal to the disulfide bonds of GPIbα generating the soluble ectodomain (glycocalicin), and leaving a remnant membrane-associated fragment that remains disulfide linked to GPIbβ. Thrombin and ADAM10/ADAM17 cleaves the ectodomain of GPV at distinct sites, with loss of GPV enhancing the capacity of thrombin to stimulate platelets via GPIbα. The stoichiometry is not shown. |
GPIbα also binds to coagulation factors from the intrinsic pathway (high molecular weight kininogen, FXII and FXI) and thrombin generated from activation of intrinsic or extrinsic coagulation pathways. Each of these factors bind to the N-terminal ligand-binding domain of GPIbα, whereas the extracellular mucin-like domain or other subunits may regulate the procoagulant activity of platelets by interacting with FVIIa and/or other factors.26,27 Activation of platelets by engagement of GPIb-IX-V or other receptors can promote coagulation by inducing the expression of anionic phospholipids on the platelet surface, thereby leading to the activation of FX to FXa and secretion of polyphosphates or other factors that promote activation of FXII.28
In addition to these interactions, other extracellular or intracellular binding partners regulate GPIb-IX-V-dependent signaling. First, the ectodomain of GPIbα interacts with platelet collagen receptor, GPVI, of the immunoreceptor family. GPVI forms a noncovalent complex with Fc receptor γ chain that is required for GPVI surface expression and for downstream following of GPVI engagement.29 GPIbα also associates with FcRγ in human platelets,30 and acts with GPVI/FcRγ as accessory signaling proteins that share common signaling pathways, as discussed below.31 Second, the intracellular domain of GPIb-IX-V interacts with calmodulin that binds to membrane-proximal amphipathic/positively charged sequences of GPIbβ and GPV and with 14-3-3θ that binds to phosphoserine-containing sequences at the C-terminal or upstream regions of GPIbα, or within GPIbβ (FIGURE 26A.2). Calmodulin dissociation regulates ectodomain shedding of GPIbα or GPV by ADAM17 or ADAM10/17, respectively.5,32,33 The regulatory p85 subunit of phosphatidyl inositol 3-kinase (PI3-kinase) also binds to the C-terminal sequence of GPIbα, independent of phosphorylation status or 14-3-3θ binding.34,35
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Molecular Basis for Platelet Secretion
Molecular Basis for Platelet Secretion
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


