INTRODUCTION
SUMMARY
Waldenström macroglobulinemia (WM) is an indolent B-cell neoplasm manifested by the accumulation of clonal immunoglobulin (Ig) M secreting lymphoplasmacytic cells. MYD88L265P and CXCR4 WHIM (warts, hypogammaglobulinemia, infections, myelokathexis)-like somatic mutations are present in more than 90 percent, and 30 to 35 percent of WM patients, respectively, and impact disease presentation, treatment outcome, and/or overall survival. Familial predisposition is common in WM. Asymptomatic patients should be observed. Patients with disease-related hemoglobin of less than 10g/dL, platelets less than 100 × 109/L, bulky adenopathy and/or organomegaly, symptomatic hyperviscosity, peripheral neuropathy, amyloidosis, cryoglobulinemia, cold-agglutinin disease, or transformed disease should be considered for therapy. Plasmapheresis should be used for patients with symptomatic hyperviscosity, and prerituximab for those with high serum IgM levels to preempt a symptomatic IgM flare. The treatment choice should take into account specific goals of therapy, necessity for rapid disease control, risk of treatment-related neuropathy, immunosuppression and secondary malignancies, and planning for future autologous stem cell transplantation. Frontline treatments include rituximab alone or combined with alkylating agents (bendamustine, cyclophosphamide), proteasome inhibitors (bortezomib, carfilzomib), or nucleoside analogues (fludarabine, cladribine). In case of relapsed or treatment-resistant patients, an alternative frontline regimen or autologous stem cell transplantation can be considered. Novel targeted agents for the treatment of WM include everolimus and ibrutinib.
Acronyms and Abbreviations
CD16, FcγRIIIa receptor; CD40L, CD40 ligand; CDR, complement determination region; CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; GM1, ganglioside M1; HCV, hepatitis C virus; Ig, immunoglobulin; IL, interleukin; κ, kappa light chain; LPL, lymphoplasmacytic lymphoma; MAG, myelin-associated glycoprotein; R-CHOP, cyclophosphamide, doxorubicin, vincristine, prednisone, and rituximab; R-CP, cyclophosphamide, prednisone, and rituximab; R-CVP, cyclophosphamide, vincristine, prednisone, and rituximab; sCD27, soluble CD27; WHO, World Health Organization; WM, Waldenström macroglobulinemia.
DEFINITION AND HISTORY
Waldenström macroglobulinemia (WM) is a lymphoid neoplasm resulting from the accumulation, predominantly in the marrow, of a clonal population of lymphocytes, lymphoplasmacytic cells, and plasma cells, which secrete a monoclonal immunoglobulin (Ig) M.1 WM corresponds to lymphoplasmacytic lymphoma (LPL) as defined in the Revised European-American Lymphoma (REAL) and World Health Organization (WHO) classification systems.2,3 Most cases of LPL are WM; less than 5 percent of cases are IgA-secreting, IgG-secreting, or nonsecreting LPL.
In 1944, Jan Waldenström, a Swedish physician-scientist, reported in Acta Medica Scandinavica three cases of a disease, he presciently thought was related to myeloma but for the absence of bone involvement and the scarcity of plasma cells in the infiltrate of small lymphocytes. He noted the increase in plasma protein concentration, marked increased serum viscosity, exaggerated bleeding and retinal hemorrhages, and virtually every other feature of the disorder in his case descriptions. In collaboration with a colleague, he showed, using ultracentrifugation and electrophoresis, that the abundant abnormal protein had a molecular weight of approximately 1 million and was not an aggregate of smaller proteins. The disease, which he described with such thoroughness, was later named in his honor.
EPIDEMIOLOGY
The age-adjusted incidence rate of WM is 3.4 per 1 million among males and 1.7 per 1 million among females in the United States. It increases in incidence geometrically with age.4,5 The incidence rate is higher among Americans of European descent. Americans of African descent represent approximately 5 percent of all patients.
Genetic factors play a role in the pathogenesis of WM. Approximately 20 percent of WM patients are of Ashkenazi-Jewish ethnic background.6 Familial disease has been reported commonly, including multigenerational clustering of WM and other B-cell lymphoproliferative diseases.7,8,9,10 Approximately 20 percent of 257 sequential patients with WM presenting to a tertiary referral center had a first-degree relative with either WM or another B-cell disorder.9 Familial clustering of WM with other immunologic disorders, including hypogammaglobulinemia and hypergammaglobulinemia (particularly polyclonal IgM), autoantibody production (particularly to the thyroid), and manifestation of hyperactive B cells has also been reported in relatives without WM.9,10 Increased expression of the BCL-2 gene with enhanced survival has been observed in B cells from familial patients and their family members.10
The role of environmental factors is uncertain, but chronic antigenic stimulation from infections and certain drug or chemical exposures have been considered but have not reached a level of scientific certainty. Hepatitis C virus (HCV) infection was implicated in WM causality in some series, but in a study of 100 consecutive WM patients in whom serologic and molecular diagnostic studies for HCV infection were performed, no association was found.11,12,13
PATHOGENESIS
Examination of the B-cell clone(s) found in the marrow of WM patients reveals a range of differentiation from small lymphocytes with large focal deposits of surface immunoglobulins, to lymphoplasmacytic cells, to mature plasma cells that contain intracytoplasmic IgM (Fig. 109–1).14 Circulating clonal B cells are often detectable in patients with WM, although lymphocytosis is uncommon.15,16 WM cells express the monoclonal IgM, and some clonal cells also express surface IgD.17 The characteristic immunophenotypic profile of WM lymphoplasmacytic cells includes the expression of the pan–B-cell markers CD19, CD20 (including FMC7), CD22, and CD79.17,18 Expression of CD5, CD10, and CD23 can be present in 10 to 20 percent of cases, and their presence does not exclude the diagnosis of WM.19 Multiparameter flow cytometric analysis has also identified CD25 and CD27 as being characteristic of the WM clone, and found that a CD22dim/CD25+/CD27+/IgM+ population may be present among clonal B lymphocytes in patients with essential monoclonal gammopathy (synonym: monoclonal gammopathy of unknown significance [MGUS]) of the IgM type who ultimately progressed to WM.20
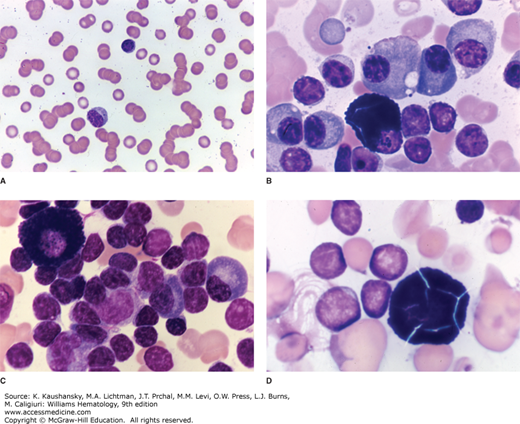
Figure 109–1.
Waldenström macroglobulinemia. A. Blood film displaying the characteristic pathologic rouleaux seen as a result of the red cell aggregating properties of immunoglobulin M. B. Marrow film showing characteristic infiltrate of lymphocytes, lymphoplasmacytic cells, and plasma cells. A mast cell is evident lower center. Although not specific for this disease, mast cells are commonly present in the marrow. C. Marrow film showing infiltrate of lymphocytes with occasional plasma cells and a mast cell. D. Marrow film showing lymphocytic infiltrate with a “cracked” mast cell sometimes seen in this disease. The fraction of plasma cells varies as shown by the somewhat higher proportion in (B) as compared to (C) and (D). Lymphocytes and lymphoplasmacytic cells predominate. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
Somatic mutations in immunoglobulin genes are present with an increased frequency of nonsynonymous as compared to silent mutations in complement determining regions, along with somatic hypermutation, thereby supporting a postgerminal center derivation for the WM B-cell clone in most patients.21,22 A strong preferential usage of VH3/JH4 gene families without intraclonal variation, and without evidence for any isotype-switched transcripts is present.23,24 These data support an IgM+ and/or IgM+IgD+ memory B-cell origin for most cases of WM.
In contrast to myeloma plasma cells, no recurrent translocations have been described in WM, which can help to distinguish WM from IgM myeloma cases, as IgM myeloma cases often exhibit t11;14 translocations.25,26 Despite the absence of IgH translocations, recurrent chromosomal abnormalities are present in WM cells. These include deletions in chromosome 6q21–23 in 40 to 60 percent of WM patients, with concordant gains in 6p in 41 percent of 6q-deleted patients.27,28,29,30 In a series of 174 untreated WM patients, 6q deletions, followed by trisomy 18, 13q deletions, 17p deletions, trisomy 4, and 11q deletions, were observed.30 Deletion of 6q and trisomy 4 were associated with adverse prognostic markers in this series. As 6q deletions represent the most recurrent cytogenetic finding in WM cases, there has been interest in identifying the region of minimal deletion and possible target genes within this region. Two putative gene candidates within this region are TNFAIP3, a negative regulator of nuclear factor kappa B signaling (NFκB), and PRDM1, a master regulator of B-cell differentiation.29,31 The removal of a NFκB-negative regulator is of particular interest as the phosphorylation and translocation of NFκB into the nucleus is a crucial event for WM cell survival.32 The success of proteasome inhibitor therapy in WM may occur because the degradation of negative regulators of NFκB, such as the inhibitor of kappa B (IκB), is blocked33,34.
A highly recurrent somatic mutation (MYD88L265P) was first identified in WM patients by whole-genome sequencing (WGS), and confirmed by multiple studies through Sanger sequencing and/or allele-specific polymerase chain reaction (PCR) assays.35,36,37,38,39,40 MYD88L265P is expressed in 90 to 95 percent of WM cases when more sensitive allele-specific PCR has been employed, using both CD19-sorted and unsorted marrow cells.36,37,38,39,40 By comparison, MYD88L265P was absent in myeloma samples, including IgM myeloma, and was expressed in a small subset (6 to 10 percent) of marginal zone lymphoma patients, who surprisingly have WM-related features.36,37,38,41 By PCR assays, 50 to 80 percent of IgM MGUS patients, also express MYD88L265P, and expression of this mutation was associated with increased risk for malignant progression.36,37,38,42 The presence of MYD88L265P in IgM MGUS patient suggests a role for this mutation as an early oncogenic driver, and other mutations and/or copy number alterations leading to abnormal gene expression are likely to promote disease progression.29
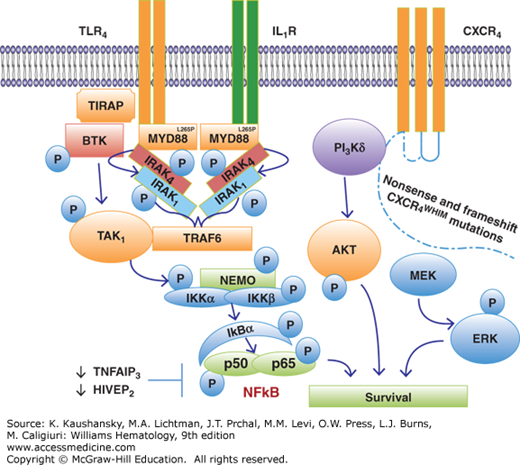
The impact of MYD88L265P to growth and survival signaling in WM cells has been addressed in several studies (Fig. 109–2). Knockdown of MYD88 decreased survival of MYD88L265P-expressing WM cells, whereas survival was enhanced by knock-in of MYD88L265P versus wild-type MYD88.43 The discovery of a mutation in MYD88 is significant given its role as an adaptor molecule in toll-like receptor (TLR) and interleukin-1 receptor (IL-1R) signaling.44 All TLRs except for TLR3 use MYD88 to facilitate their signaling. Following TLR or IL-1R stimulation, MYD88 is recruited to the activated receptor complex as a homodimer which then complexes with IRAK4 and activates IRAK1 and IRAK2.45,46,47 Tumor necrosis factor receptor–associated factor 6 is then activated by IRAK1 leading to NFκB activation via IκBα phosphorylation.48 Use of inhibitors of MYD88 pathway led to decreased IRAK1 and IκBα phosphorylation, as well as survival of MYD88L265P expressing WM cells. These observations are of particular relevance to WM since NFκB signaling is important for WM growth and survival.49 Bruton tyrosine kinase (BTK) is also activated by MYD88L265P.43 Activated BTK coimmunoprecipitates with MYD88 that could be abrogated by use of a BTK kinase inhibitor, and overexpression of MYD88L265P, but not wild-type(WT) MYD88, triggers BTK activation. Knockdown of MYD88 by lentiviral transfection or use of a MYD88 homodimerization inhibitor also abrogated BTK activation in MYD88L265P-mutated WM cells.
Figure 109–2.
MYD88L265P and CXCR4WHIM mutations are highly prevalent in patients with Waldenström macroglobulinemia, and trigger transcriptional factors that include nuclear factor κB (NFκB), AKT, and extracellular signal-regulated kinase (ERK) that support the growth and survival of lymphoplasmacytic cells.
The second most common somatic mutation after MYD88L265P revealed by WGS was found in the C-terminus of the CXCR4 receptor. These mutations are present in 30 to 35 percent of WM patients, and impact serine phosphorylation sites that regulate CXCR4 signaling by its only known ligand, stromal cell-derived factor (SDF)-1a (CXCL12).29,50,51,52 The location of somatic mutations found in the C-terminus of CXCR4 in WM are similar to those observed in the germline of patients with WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis) syndrome, a congenital immunodeficiency disorder characterized by chronic noncyclic neutropenia.53 Patients with WHIM syndrome exhibit impaired CXCR4 receptor internalization following SDF-1a stimulation, which results in persistent CXCR4 activation and myelokathexis.54
In WM patients, two classes of CXCR4 mutations occur in the C-terminus. These include nonsense (CXCR4WHIM/NS) mutations, which truncate the distal 15- to 20-amino-acid region, and frameshift (CXCR4WHIM/FS) mutations, which compromise a region of up to 40 amino acids in the C-terminal domain.29,50 Nonsense and frameshift mutations are almost equally divided among WM patients with CXCR4 somatic mutations, and more than 30 different types of CXCR4WHIM mutations have been identified in WM patients.29,50 Preclinical studies with WM cells engineered to express nonsense and frameshift CXCR4WHIM-mutated receptors have shown enhanced and sustained AKT and extracellular signal-regulated kinase (ERK) signaling following SDF-1a relative to CXCR4WT (see Fig. 109–2), as well increased cell migration, adhesion, growth and survival, and drug resistance of WM cells.51,55,56
Many copy number alterations have been revealed in WM patients that impact growth and survival pathways. Frequent loss of HIVEP2 (80 percent) and TNFAIP3 (50 percent) genes that are negative regulators of NFkB expression (Fig. 109–2), as well as LYN (70 percent) and IBTK (40 percent) that modulate B-cell receptor (BCR) signaling have been revealed by WGS.29 WGS has also revealed common defects in chromatin remodeling with somatic mutations in ARID1A present in 17 percent, and loss of ARID1B in 70 percent of WM patients. Both ARID1A and ARID1B are members of the SWI/SNF family of proteins, and are thought to exert their effects via p53 and CDKN1A regulation. TP53 is mutated in 7 percent of sequenced WM genomes, while PRDM2 and TOP1 that participate in TP53-related signaling are deleted in 80 percent and 60 percent of WM patients, respectively.29 Taken together, somatic events that contribute to impaired DNA damage response are also common in WM.
MYD88 and CXCR4 mutations are important determinants of the clinical presentation of WM patients. Significantly higher marrow disease involvement, serum IgM levels, and symptomatic disease requiring therapy, including hyperviscosity syndrome was observed in those patients with MYD88L265PCXCR4WHIM/NS mutations.50 Patients with MYD88L265PCXCR4WHIM/FS or MYD88L265PCXCR4WT had intermediate marrow and serum IgM levels; those with MYD88WTCXCR4WT showed the lowest marrow disease burden. Fewer patients with MYD88L265P and CXCR4WHIM/FS or CXCR4WHIM/NS, compared to MYD88L265PCXCR4WT presented with adenopathy, further delineating differences in disease tropism based on CXCR4 status. Despite the more-aggressive presentation associated with CXCR4WHIM/NS genotype, risk of death was not impacted by CXCR4 mutation status. Risk of death was found to be 10-fold higher in patients with MYD88WT versus MYD88L265P genotype.50
Increased numbers of mast cells are found in the marrow of WM patients, wherein they are usually admixed with tumor cell aggregates (see Fig. 109–1).14,18,57 The role of mast cells in WM was investigated in one study wherein coculture of primary autologous or mast cell lines with WM cells resulted in dose-dependent WM cell proliferation and/or tumor colony formation, through CD40 ligand (CD40L) signaling.57 WM cells release soluble CD27 (sCD27), which may be triggered by cleavage of membrane-bound CD27 by matrix metalloproteinase 8 (MMP8).58 sCD27 levels are elevated in the serum of WM patients, and follow disease burden in mice engrafted with WM cells, as well as in WM patients.60 sCD27 triggers the upregulation of CD40L and a proliferation-inducing ligand (APRIL) on mast cells derived from WM patients, and mast cell lines through its receptor CD70. Modeling in mice engrafted with a CD70-blocking antibody shows inhibition of tumor cell growth, suggesting that WM cells require a microenvironmental support system for their growth and survival.59 High levels of CXCR4 and very late antigen-4 (VLA-4) have also been observed in WM cells.60 In blocking experiments studies, CXCR4 supported migration of WM cells, while VLA-4 contributed to adhesion of WM cells to marrow stromal cells.60
CLINICAL FEATURES
Table 109–1 presents the clinical and laboratory findings at time of diagnosis of WM in one large institutional study.16 Unlike most indolent lymphomas, splenomegaly and lymphadenopathy are uncommon (≤15 percent). Purpura is frequently associated with cryoglobulinemia and in rare circumstances with light-chain (AL) amyloidosis (Chap. 108). Hemorrhagic and neuropathic manifestations are multifactorial (see “Immunoglobulin M–Related Neuropathy” below). The morbidity associated with WM is caused by the concurrence of two main components: tissue infiltration by neoplastic cells and, importantly, the physicochemical and immunologic properties of the monoclonal IgM. As shown in Table 109–2, the monoclonal IgM can produce clinical manifestations through several different mechanisms related to its physicochemical properties, nonspecific interactions with other proteins, antibody activity, and tendency to deposit in tissues.61,62,63
| Median | Range | Normal Reference Range | |
|---|---|---|---|
| Age (years) | 58 | 32–91 | NA |
| Gender (male/female) | 215/141 | NA | |
| Marrow involvement (% of area on slide) | 30 | 5–95 | NA |
| Adenopathy (% of patients) | 15 | NA | |
| Splenomegaly (% of patients) | 10 | NA | |
| IgM (mg/dL) | 2620 | 270–12,400 | 40–230 |
| IgG (mg/dL) | 674 | 80–2770 | 700–1600 |
| IgA (mg/dL) | 58 | 6–438 | 70–400 |
| Serum viscosity (cp) | 2.0 | 1.1–7.2 | 1.4–1.9 |
| Hematocrit (%) | 35 | 17–45 | 35–44 |
| Platelet count (× 109/L) | 275 | 42–675 | 155–410 |
| White cell count (× 109/L) | 6.4 | 1.7–22 | 3.8–9.2 |
| β2M (mg/dL) | 2.5 | 0.9–13.7 | 0–2.7 |
| LDH (U/mL) | 313 | 61–1701 | 313–618 |
| Properties of IgM Monoclonal Protein | Diagnostic Condition | Clinical Manifestations |
|---|---|---|
| Pentameric structure | Hyperviscosity | Headaches, blurred vision, epistaxis, retinal hemorrhages, leg cramps, impaired mentation, intracranial hemorrhage |
| Precipitation on cooling | Cryoglobulinemia (type I) | Raynaud phenomenon, acrocyanosis, ulcers, purpura, cold urticaria |
| Autoantibody activity to myelin-associated glycoprotein, ganglioside M1, sulfatide moieties on peripheral nerve sheaths | Peripheral neuropathies | Sensorimotor neuropathies, painful neuropathies, ataxic gait, bilateral foot drop |
| Autoantibody activity to IgG | Cryoglobulinemia (type II) | Purpura, arthralgia, renal failure, sensorimotor neuropathies |
| Autoantibody activity to red blood cell antigens | Cold agglutinins | Hemolytic anemia, Raynaud phenomenon, acrocyanosis, livedo reticularis |
| Tissue deposition as amorphous aggregates | Organ dysfunction | Skin: bullous skin disease, papules, Schnitzler syndrome |
| Gastrointestinal: diarrhea, malabsorption, bleeding | ||
| Kidney: proteinuria, renal failure (light-chain component) | ||
| Tissue deposition as amyloid fibrils (light-chain component most commonly) | Organ dysfunction | Fatigue, weight loss, edema, hepatomegaly, macroglossia, organ dysfunction of involved organs (heart, kidney, liver, peripheral sensory and autonomic nerves) |
The increased plasma IgM levels leads to blood hyperviscosity and its complications.64 The mechanisms behind the marked increase in the resistance to blood flow and the resulting impaired transit through the microcirculatory system are complex.64,65,66,67 The main determinants are (1) a high concentration of monoclonal IgMs, which may form aggregates and may bind water through their carbohydrate component; and (2) their interaction with blood cells. Monoclonal IgM increases red cell aggregation (rouleaux formation; see Fig. 109–1) and red cell internal viscosity while reducing red cell deformability. The presence of cryoglobulins contributes to increasing blood viscosity, as well as to the tendency to induce erythrocyte aggregation. Serum viscosity is proportional to IgM concentration up to 30 g/L, then increases sharply at higher levels. Increased plasma viscosity may also contribute to inappropriately low erythropoietin production, which is the major reason for anemia in these patients.67 Renal synthesis of erythropoietin is inversely correlated with plasma viscosity. Clinical manifestations are related to circulatory disturbances that can be best appreciated by ophthalmoscopy, which shows distended and tortuous retinal veins, hemorrhages, and papilledema (Fig. 109–3).68 Symptoms usually occur when the monoclonal IgM concentration exceeds 50 g/L or when serum viscosity is greater than 4.0 centipoises (cp), but there is individual variability, with some patients showing no evidence of hyperviscosity even at 10 cp.64 The most common symptoms are oronasal mucosal bleeding, visual disturbances because of retinal bleeding, and dizziness that rarely may lead to stupor or coma. Heart failure can be aggravated, particularly in the elderly, owing to increased blood viscosity, expanded plasma volume, and anemia. Inappropriate red cell transfusion can exacerbate hyperviscosity and may precipitate cardiac failure.
Figure 109–3.
Funduscopic examination of a patient with Waldenström macroglobulinemia with hyperviscosity-related changes, including dilated retinal vessels, hemorrhages, and “venous sausaging.” The white material at the edge of the veins may be cryoglobulin. (Used with permission of Marvin J. Stone, MD.)

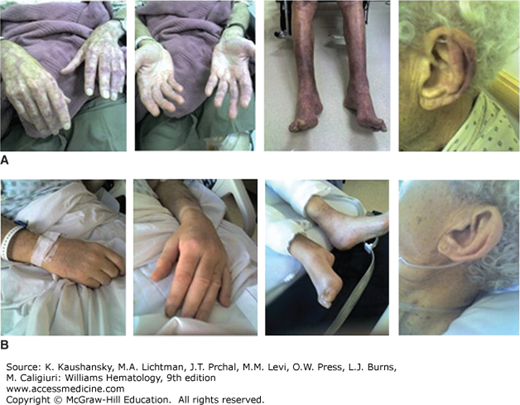
The monoclonal IgM can behave as a cryoglobulin in up to 20 percent of patients, and is usually type I and asymptomatic in most cases.16,64,70 Cryoprecipitation is mainly dependent on the concentration of monoclonal IgM; for this reason plasmapheresis or plasma exchange are commonly effective in this condition. Symptoms result from impaired blood flow in small vessels and include Raynaud phenomenon, acrocyanosis, and necrosis of the regions most exposed to cold, such as the tip of the nose, ears, fingers, and toes (Fig. 109–4), malleolar ulcers, purpura, and cold urticaria. Renal manifestations are infrequent. Mixed cryoglobulins (type II) consisting of IgM–IgG complexes may be associated with hepatitis C infections.70
Monoclonal IgM may exert its pathogenic effects through specific recognition of autologous antigens, the most notable being nerve constituents, immunoglobulin determinants, and red blood cell antigens.
IgM-related peripheral neuropathy is common in WM patients, with estimated prevalence rates of 5 to 40 percent.71,72,73 Approximately 8 percent of idiopathic neuropathies are associated with a monoclonal gammopathy, with a preponderance of IgM (60 percent) followed by IgG (30 percent) and IgA (10 percent) (Chap. 106).74,75 The nerve damage is mediated by diverse pathogenetic mechanisms: (1) IgM antibody activity toward nerve constituents causing demyelinating polyneuropathies; (2) endoneurial granulofibrillar deposits of IgM without antibody activity, associated with axonal polyneuropathy; (3) occasionally by tubular deposits in the endoneurium associated with IgM cryoglobulin; and, rarely, (4) by amyloid deposits or by neoplastic cell infiltration of nerve structures.73,76 Half of the patients with IgM neuropathy have a distinctive clinical syndrome that is associated with antibodies against a minor 100-kDa glycoprotein component of nerve known as the myelin-associated glycoprotein (MAG). Anti-MAG antibodies are generally monoclonal IgMκ, and usually also exhibit reactivity with other glycoproteins or glycolipids that share antigenic determinants with MAG.77,78,79 The anti–MAG-related neuropathy is typically distal and symmetrical, affecting both motor and sensory functions; it is slowly progressive with a long period of stability.72,80 Most patients present with sensory complaints (paresthesias, aching discomfort, dysesthesias, or lancinating pains), imbalance and gait ataxia, owing to lack proprioception; leg muscles atrophy in advanced stage. Patients with predominantly demyelinating sensory neuropathy in association with monoclonal IgM to gangliosides with disialosyl moieties, such as GD1b, GD3, GD2, GT1b, and GQ1b, have also been reported.81,82 Anti-GD1b and anti-GQ1b antibodies were associated with sensory ataxic neuropathy. These antiganglioside monoclonal IgMs present core clinical features of chronic ataxic neuropathy sometimes with present ophthalmoplegia and/or red blood cell cold agglutinating activity. The disialosyl epitope is also present on red blood cell glycophorins, thereby accounting for the red cell cold agglutinin activity of anti-Pr2 specificity.83,84 Monoclonal IgM proteins that bind to gangliosides with a terminal trisaccharide moiety, including ganglioside M2 (GM2) and GalNac-GD1A, are associated with chronic demyelinating neuropathy and severe sensory ataxia, unresponsive to glucocorticoids.85 Antiganglioside IgM proteins may also cross-react with lipopolysaccharides of Campylobacter jejuni, whose infection is known to precipitate the Miller-Fisher syndrome, a variant of the Guillain-Barré syndrome.86 Thus, molecular mimicry may play a role in this condition. Antisulfatide monoclonal IgM proteins, associated with sensory-sensorimotor neuropathy, have been detected in 5 percent of patients with IgM monoclonal gammopathy and neuropathy.87 Motor neuron disease has been reported in patients with WM and monoclonal IgM with anti-GM1 and sulfoglucuronyl paragloboside activity.88 Polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes (the POEMS syndrome) are rare in patients with WM.89
Monoclonal IgM may have cold agglutinin activity, that is, it can recognize specific red cell antigens at temperatures below 37°C, producing chronic hemolytic anemia. This disorder occurs in less than 10 percent of WM patients and is associated with cold agglutinin titers greater than 1:1000 in most cases.90 The monoclonal component is usually an IgMκ and reacts most commonly with red cell I/i antigens, resulting in complement fixation and activation.91,92 Mild to moderate chronic hemolytic anemia can be exacerbated after cold exposure. Hemoglobin usually remains above 70 g/L. The hemolysis is usually extravascular, mediated by removal of C3b opsonized red cells by the mononuclear phagocyte system, primarily in the liver. Intravascular hemolysis from complement destruction of red blood cell membrane is infrequent. The agglutination of red cells in the skin circulation also causes Raynaud syndrome, acrocyanosis, and livedo reticularis. Macroglobulins with the properties of both cryoglobulins and cold agglutinins with anti-Pr specificity can occur. These properties may have as a common basis the binding of the sialic acid-containing carbohydrate present on red blood cell glycophorins and on Ig molecules. Several other macroglobulins with antibody activity toward autologous antigens (e.g., phospholipids, tissue and plasma proteins) and foreign ligands have also been described.
The monoclonal protein can deposit in several tissues as amorphous aggregates. Linear deposition of monoclonal IgM along the skin basement membrane is associated with bullous skin disease.93 Amorphous IgM deposits in the dermis result in IgM storage papules on the extensor surface of the extremities, referred to as macroglobulinemia cutis.94 Deposition of monoclonal IgM in the lamina propria and/or submucosa of the intestine may be associated with diarrhea, malabsorption, and gastrointestinal bleeding.95,96 Kidney involvement is less common and less severe in WM than in myeloma, probably because the amount of light chain excreted in the urine is generally lower in WM than in myeloma and because of the absence of contributing factors, such as hypercalcemia. Urinary cast nephropathy, however, has occurred in WM.97 On the other hand, the IgM macromolecule is more susceptible to being trapped in the glomerular loops where ultrafiltration presumably contributes to its precipitation, forming subendothelial deposits of aggregated IgM proteins that occlude the glomerular capillaries.98 Mild and reversible proteinuria may result and most patients are asymptomatic. The deposition of monoclonal light chain as fibrillar amyloid deposits (AL amyloidosis) is uncommon in patients with WM.99 Clinical expression and prognosis are similar to those of other AL amyloidosis patients with involvement of heart (44 percent), kidneys (32 percent), liver (14 percent), lungs (10 percent), peripheral or autonomic nerves (38 percent), and soft tissues (18 percent). The incidence of cardiac and pulmonary involvement is higher in patients with monoclonal IgM than with other immunoglobulin isotypes. The association of WM with reactive amyloidosis has been documented rarely.100,101 Simultaneous occurrence of fibrillary glomerulopathy, characterized by glomerular deposits of wide noncongophilic fibrils and amyloid deposits, has been described.102
Tissue infiltration by neoplastic cells is uncommon but can involve various organs and tissues, including the liver, spleen, lymph nodes, lungs, gastrointestinal tract, kidneys, skin, eyes, and central nervous system.
Pulmonary involvement in the form of masses, nodules, diffuse infiltrate, or pleural effusions is uncommon; the overall incidence of pulmonary and pleural findings is approximately 4 percent.103,104,105 Cough is the most common presenting symptom, followed by dyspnea and chest pain. Chest radiographic findings include parenchymal infiltrates, confluent masses, and effusions.
Malabsorption, diarrhea, bleeding, or obstruction may indicate involvement of the gastrointestinal tract at the level of the stomach, duodenum, or small intestine.106,107,108,109
In contrast to myeloma, infiltration of the kidney interstitium with lymphoplasmacytoid cell can occur in WM, and renal or perirenal masses are not uncommon.110,111
The skin can be the site of dense lymphoplasmacytic infiltrates, similar to that seen in the liver, spleen, and lymph nodes, forming cutaneous plaques and, rarely, nodules.112 Chronic urticaria and IgM gammopathy are the two cardinal features of the Schnitzler syndrome, which is not usually associated initially with clinical features of WM, although evolution to WM is not uncommon.113 Thus, close followup of these patients is important.
Invasion of articular and periarticular structures by WM malignant cells is rarely reported.114
The neoplastic cells can infiltrate the periorbital structures, lacrimal gland, and retroorbital lymphoid tissues, resulting in ocular nerve palsies.115,116
Direct infiltration of the central nervous system by monoclonal lymphoplasmacytic cells as infiltrates or as tumors constitutes the rarely observed Bing-Neel syndrome, characterized clinically by confusion, memory loss, disorientation, and motor dysfunction (reviewed in Ref. 117).
LABORATORY FINDINGS
Anemia is the most common finding in patients with symptomatic WM and is caused by a combination of factors: decrease in red cell survival, impaired erythropoiesis, moderate plasma volume expansion, hepcidin production leading to iron reutilization defect, and blood loss from the gastrointestinal tract.16,118,119 Blood films are usually normocytic and normochromic, and rouleaux formation is often pronounced (see Fig. 109–1). Mean red cell volume may be elevated spuriously owing to erythrocyte aggregation. In addition, the hemoglobin estimate can be inaccurate, that is, falsely high, because of interaction between the monoclonal protein and the diluent used in some automated analyzers.120
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


