Lymphoproliferative Disorders
Elizabeth Margolskee, MD, MPH
Peter Martin, MD
The diagnosis and classification of lymphoid tumors is a multistep process involving integration of clinical, morphologic, phenotypic, and molecular data. The first step in the evaluation of pathologic specimens for lymphoma is distinguishing benign from malignant lymphoproliferations. Generally, lymphomas are characterized by distortion of the normal nodal or tissue architecture, a monotonous-appearing cellular proliferation, and “atypical” features, like necrosis or a high mitotic rate. However, some indolent lymphoproliferations can be quite subtle to identify morphologically; in these cases, integration of clinical and morphologic data with flow cytometry and other molecular testing may help establish a diagnosis. Once the diagnosis of a lymphoma is made, the World Health Organization (WHO) classification provides a framework for subtyping lymphoid neoplasms into three major divisions: mature B-cell lymphoma, mature T/natural killer (NK)-cell lymphoma, and Hodgkin lymphoma. About 85% of lymphoid neoplasms are of B-cell origin; nearly all the rest derive from T cells. Tumors arising from NK cells are rare. A complete discussion of this classification is beyond the scope of this text; this chapter will briefly review features typical of the most common lymphoproliferative disorders.
MATURE B-CELL LYMPHOMA
B-Cell Ontogeny and the Mature B-Cell Neoplasms
The entire B-cell line originates from precursor B lymphoblasts in the bone marrow that differentiate into mature B cells expressing surface immunoglobulin. The lymphoblasts are the putative cells of origin of precursor B acute lymphoblastic leukemia and lymphoma. The naïve marrow B cells are the cells of origin of some chronic lymphocytic leukemias (CLLs)/small lymphocytic lymphomas (SLLs). Immature B cells exit the bone marrow and undergo variable (V), diversity (D), and joining (J) gene segment (VDJ) recombination of immunoglobulin genes, differentiating into naïve B cells. The naïve B cells leave the marrow to circulate in the blood and travel to the cortex of lymph nodes, where they occupy primary follicles (those without germinal centers) and secondary follicles (those with germinal centers) in the mantle zone, which surrounds the germinal centers. These cells are the source of mantle cell lymphoma (MCL). When naïve B cells encounter antigen, they transform into blasts and travel to the center of primary follicles, forming the germinal center, where the cells are called centroblasts. These large cells, whose vesicular nuclei contain nucleoli, are thought to be the source of most large B-cell lymphomas and Burkitt lymphoma. The centroblasts mature to centrocytes, which are medium-sized, cleaved cells with inconspicuous nucleoli from which follicular lymphomas are thought to arise. The centrocytes eventually leave the germinal center and undergo plasmacytic differentiation. It is these cells that give rise to the activated B-cell subtype of diffuse large B-cell lymphoma. Post-germinal center B cells differentiate further into: (1) antibody-secreting plasma cells, from which plasma cell myeloma and Waldenström macroglobulinemia originate, and (2) memory B cells, which are found in the areas around follicles (marginal zone) and are the origin of marginal zone lymphomas of the spleen, lymph nodes, and mucosa-associated lymphoid tissue ( MALT). The origin of cells in mature B-cell neoplasms ranges in differentiation from naïve B cells—resting lymphoid cells that have surface immunoglobulin but have not yet encountered antigens—to mature plasma cells, which produce immunoglobulins in response to previous antigen exposure. The classification of these disorders, which account for >80% of lymphoid neoplasms, generally depends on morphologic characteristics, immunophenotyping of the cells, and anatomic involvement (i.e., whether the process is primarily disseminated [often leukemic], extranodal, or nodal). In addition, molecular diagnostics (e.g., karyotype, fluorescent in situ hybridization [FISH] studies, and specific point mutations) play a role
in lymphoma diagnosis. In this chapter, the mature B-cell neoplasms have been organized by cell of origin to reflect the role of B-cell ontogeny in lymphomagenesis.
in lymphoma diagnosis. In this chapter, the mature B-cell neoplasms have been organized by cell of origin to reflect the role of B-cell ontogeny in lymphomagenesis.
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
CLL/SLL is a clinically heterogeneous disease consisting of monoclonal small B-lymphocytes expressing CD19, CD5, and CD23. The presentation can be predominantly leukemic with bone marrow involvement (i.e., CLL) or may present in lymph nodes without a significant leukemic component (i.e., SLL). Most patients are asymptomatic at the time of diagnosis, their median age is about 65, and the male:female ratio is approximately 2:1. When symptoms occur, they commonly include fatigue related to anemia from bone marrow replacement, splenomegaly, or immune-related hemolysis caused by a warm-reactive polyclonal IgG; the latter occurs in about 10% to 25% of patients during the course of disease.
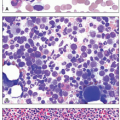
CLL is characterized by a monoclonal B-cell lymphocytosis in peripheral blood of at least 5 × 109/L with a specific immunophenotype: CD5 and CD23 positivity with low levels of surface immunoglobulin and CD20. The blood smear shows an increased number of mature small lymphocytes with little cytoplasm and dense, clumped chromatin. Nucleoli are not usually visible, and many cells, being more fragile than normal lymphocytes, disrupt during the preparation of the smear, producing “smudge cells,” in which the cytoplasm is lost and the nucleus spreads out. Some cells may be prolymphocytes, which are larger than mature lymphocytes, possess nucleoli, and have more cytoplasm. The presence of a small circulating CD5-positive monoclonal B-cell population (i.e., <5 × 109/L) in the absence of lymphadenopathy is termed monoclonal B-cell lymphocytosis (MBL), which precedes the development of virtually all cases of CLL. MBL is seen in 12% of healthy individuals and is divided into “high count” and “low count” at a threshold of 0.5 × 109/L. Individuals with “low count” MBL have little risk of disease; “high count” MBL should be followed for progression to CLL. Disease prognosis varies by immunophenotype, cytogenetic findings, and mutational status. Analysis of the immunoglobulin heavy chain gene for somatic hypermutation has demonstrated that CLL consists of two subsets: an unmutated form and a mutated form. Unmutated CLL arises from naïve B cells and mutated CLL arises from memory B cells. In the immunochemotherapy era, the presence of somatic hypermutation in the IgH gene was associated with good prognosis, whereas expression of CD38, ZAP-70, and CD49d were markers of an aggressive course. Deletion of 13q is a favorable cytogenetic finding, whereas del(17p), del(11q), and del(6q) tend to indicate poor prognosis. The molecular landscape of CLL/SLL is heterogeneous, with mutations in TP53, NOTCH1, SF3B1, and BIRC3 frequently seen and associated with a worse prognosis. As therapeutic options have evolved to include B-cell receptor antagonists (e.g., inhibitors of Bruton tyrosine kinase or phosphoinositol triphosphate kinase), the predictive value of these markers is also in flux and is the subject of active study.
About 5% of patients with CLL/SLL develop Richter syndrome. The CLL/SLL transforms into a non-Hodgkin lymphoma, usually diffuse large B-cell type. Characteristic features of Richter syndrome include fever, weight loss, rapid increase in lymph node size, and elevated serum lactic dehydrogenase (LDH). Patients with transformed CLL should be reevaluated for signs of progression, including genetic or karyotypic evolution.
B-Cell Prolymphocytic Leukemia
This rare disorder predominantly affects older adults (median age, 70), with a male:female ratio of 1.6:1. Most patients have marked splenomegaly and lymphocytosis without enlarged peripheral lymph nodes. By definition, prolymphocytes constitute over 55% of the circulating lymphoid cells, but in most cases they exceed 90%. About half of the patients have anemia and thrombocytopenia. The prolymphocytes are twice the size of small lymphocytes and possess a small amount of pale blue cytoplasm and round nuclei, which contain moderately condensed chromatin and a conspicuous central nucleolus. The bone marrow shows diffuse infiltration with these abnormal cells.
Mantle Cell Lymphoma
This lymphoma, which constitutes about 4% to 5% of non-Hodgkin lymphoma, occurs primarily in adults, with a median age at diagnosis of about 60 years and a male predominance of at least 2:1. It is thought to arise from naïve B cells normally present in the mantle zone and develops along two distinct pathways. Classical MCL involves lymph nodes and other extranodal sites, has unmutated immunoglobulin heavy chain variable genes, IGHV, and may be aggressive. In contrast, leukemic non-nodal MCL is limited to the bone marrow, peripheral blood, and spleen, has mutated IGHV, and is generally indolent. The neoplastic cell closely resembles centrocytes, appearing as small- to medium-sized lymphocytes with sparse cytoplasm and irregular or cleaved nuclei containing moderately dispersed chromatin and inconspicuous nucleoli. In the blastoid variant of classical MCL, the neoplastic cells appear enlarged with a fine chromatin pattern. Bone marrow involvement in MCL includes interstitial, focal, paratrabecular, and diffuse patterns. The prognosis of MCL is slightly worse than the other small mature B-cell lymphomas; individuals with the blastoid variant fare worst, with a median overall survival of slightly more than a year.
Most cases of MCL are driven by a translocation of cyclin D1 and the immunoglobulin heavy chain gene, which can be detected by immunohistochemical testing for cyclin D1 or by FISH studies. A recently described subset of MCLs is negative for cyclin D1; these cases generally express SOX-11.
Follicular Lymphoma
Follicular lymphoma constitutes about 20% of all lymphoma in the United States, primarily affecting adults, with a median age of about 60 years and equal gender distribution. Most patients have widespread disease at diagnosis, with diffuse
lymph node enlargement, bone marrow involvement in about 40% of patients, and circulating neoplastic cells in about 10%. In 25% to 35% of cases, at a rate of 1% to 3% per year, the disease transforms into a large B-cell lymphoma, usually diffuse.
lymph node enlargement, bone marrow involvement in about 40% of patients, and circulating neoplastic cells in about 10%. In 25% to 35% of cases, at a rate of 1% to 3% per year, the disease transforms into a large B-cell lymphoma, usually diffuse.
Follicular lymphoma arises from germinal center B lymphocytes; these cells recapitulate the constituency of normal lymphoid follicles: centrocytes and centroblasts. The centrocytes are small with cleaved or otherwise irregular nuclear contours. Centroblasts are larger with oval nuclei with open chromatin and prominent nucleoli. Histologic examination of the lymph node generally shows back-to-back follicles, which are composed entirely of neoplastic cells. The proportion of centroblasts then determines the grade: grade 1 or 2 (low grade) has 0 to 15 centroblasts per high-power field (hpf); grade 3a has more than 15 centroblasts/hpf with centrocytes also present; grade 3b has more than 15 centroblasts/hpf without centrocytes. Thus, grade 3b can often be difficult to distinguish from diffuse large B-cell lymphoma. In peripheral blood smears, the neoplastic cells are commonly smaller than normal lymphocytes, have very sparse cytoplasm, and possess cleft nuclei. Bone marrow involvement is generally paratrabecular and is composed primarily of centrocytes. Follicular lymphoma is characterized by a rearrangement of BCL2 with the immunoglobulin heavy chain gene in 80% to 90% of cases, resulting in a translocation of chromosomes 14 and 18 and expression of BCL2 by immunohistochemistry (IHC). Markers of germinal center cell origin, such as CD10, BCL6, LMO2, and HGAL, are also positive in follicular lymphoma.
Diffuse Large B-Cell Lymphoma
The large B-cell lymphomas are defined by lymphoid cells with nuclei that are twice the size of a small lymphocyte. There are numerous distinct variants of large B-cell lymphoma, many of which are vanishingly rare. Diffuse large B-cell lymphoma accounts for 25% of non-Hodgkin lymphoma and is generally a disease of the elderly. Patients commonly present with a rapidly enlarging mass, which can be in virtually any site. Bone marrow involvement, however, is rare. The proliferation is generally diffuse, with obliteration of normal follicular dendritic meshworks.
Diffuse large B-cell lymphoma is a heterogeneous group and can arise from B cells at different stages of maturation. Gene expression profiling studies of diffuse large B-cell lymphoma have identified two distinct subgroups with an expression profile characteristic of either normal germinal center cells (GCB-like) or activated blood memory B cells (ABC-like) and have demonstrated that patients with the ABC subtype have a worse prognosis when treated with immunochemotherapy. IHC, including the Hans algorithm, which relies on expression of CD10, BCL6, and MUM1, can be used to identify GCB and non-GC subgroups with reasonable accuracy, although the prognostic value of IHC-defined cell of origin remains unclear. Expression of MYC and BCL2 are commonly evaluated by IHC, and positivity for both may be associated with poor outcomes. Cytogenetic analysis for rearrangements of MYC, BCL2, and BCL6 is commonly performed (see below). A subset of DLBCL is Epstein-Barr virus (EBV) driven (EBV+ DLBCL, NOS) and may be associated with some degree of immunosuppression.
High-Grade B-Cell Lymphoma
This category encompasses large B-cell lymphomas with either morphologic or molecular features that are thought to indicate poor prognosis. There are two subgroups that compose this category: (1) Large B-cell lymphomas with blastoid or Burkitt-like features may be classified as high-grade B-cell lymphoma, not otherwise specified, although at present, there is little consensus about what morphologic features indicate more aggressive disease; or (2) large B-cell lymphomas with MYC and either BCL2 or BCL6 rearrangements, known historically as “double-hit lymphoma,” also fall into this category as high-grade B-cell lymphoma, with rearrangements of MYC and BCL2 and/or BCL6.
Mediastinal Large B-Cell Lymphoma
This subtype of diffuse large B-cell lymphoma commonly affects young women and presents with low stage, bulky mediastinal disease, causing airway obstruction. Histologically, the B cells are large and embedded in a background of prominent sclerosis. MYC, BCL2, and BCL6 gene rearrangements are uncommon.
Primary Effusion Lymphoma
This subtype of large B-cell lymphoma was originally identified in the early days of the human immunodeficiency virus (HIV) epidemic. Most cases arise in immunocompromised hosts (HIV most commonly, but also organ transplant recipients and elderly individuals) and, while they tend to involve body cavities (e.g., pleural, pericardial, or peritoneal), rare extracavitary “solid” cases of primary effusion lymphoma have been reported in various nodal and extranodal sites. It is universally associated with HHV8 infection and a subset of cases shows coinfection with EBV. The neoplastic cells are large and plasmablastic with eccentric nuclei, a perinuclear clear zone, and prominent nucleoli. The cytoplasm is basophilic and may contain vacuoles. These cells express CD45 and plasma cell markers (CD138, CD38, CD30). They typically do not express B- or T-cell antigens or surface immunoglobulin. Immunostaining for LANA, an HHV8-associated antigen, can be useful in diagnosis.
Burkitt Lymphoma
Burkitt lymphoma is the most common form of non-Hodgkin lymphoma in children; however, the majority of cases arise in adults. It occurs in three distinct epidemiologic settings. The endemic form is EBV-driven and affects young children from equatorial Africa and Papua New Guinea. Common sites of involvement include the bones of the face, gonads, and kidneys. The sporadic form occurs worldwide, predominantly arising in extranodal sites in children and young adults. The
minority of cases are associated with EBV. The immunodeficiency-associated form of Burkitt lymphoma can be seen not only in HIV-positive individuals, but also in patients with iatrogenic immunosuppression. In immunodeficient hosts, Burkitt lymphoma more frequently involves the bone marrow and lymph nodes.
minority of cases are associated with EBV. The immunodeficiency-associated form of Burkitt lymphoma can be seen not only in HIV-positive individuals, but also in patients with iatrogenic immunosuppression. In immunodeficient hosts, Burkitt lymphoma more frequently involves the bone marrow and lymph nodes.
Burkitt lymphoma is classically described as having a “starry sky” appearance, with sheets of dark neoplastic lymphocytes punctuated by scattered benign histiocytes with clear cytoplasm. The neoplastic, uniform-appearing cells have vacuolated cytoplasm, clumped chromatin, and often multiple nucleoli. The classic immunophenotype is positive for CD10 and BCL6 with no expression of BCL2 or TdT. The Ki-67 proliferation index should be almost 100%. MYC rearrangements are seen in all cases, most commonly with the Ig heavy chain locus on chromosome 14, or rarely with the κ or λ light chain loci on chromosomes 2 or 22, respectively. The diagnosis of Burkitt lymphoma can be difficult in cases with atypical morphology or an aberrant immunophenotype.
Marginal Zone Lymphoma
Post-germinal center memory B cells give rise to marginal zone lymphoma, which is further subclassified based on the sites of involvement into splenic, nodal, and extranodal (MALT) lymphoma. Marginal zone lymphomas generally lack CD5, CD10, and CD23 expression; at present, sensitive and specific markers are an area of active research. The diagnosis tends to be one of exclusion. Thus, although splenic, nodal, and extranodal marginal zone lymphoma share a common cell of origin, they have distinct epidemiologic, pathologic, and molecular features.
Splenic Marginal Zone Lymphoma
This rare disorder, previously called splenic lymphoma with villous lymphocytes, is a B-cell neoplasm involving the splenic white pulp and bone marrow with a small component of circulating neoplastic cells. The splenic white pulp architecture, which normally consists of reactive germinal centers, is altered by a proliferation of larger “monocytoid” B cells in the marginal zone. The germinal centers are colonized by small centrocyte-like cells with some extension into the red pulp. Most patients are over 50 years old, and both genders are equally affected. Hepatitis C infection has been associated with splenic marginal zone lymphoma (SMZL), and hepatitis C virus-related SMZLs may respond to antiviral therapy. The major clinical feature is splenomegaly and hypersplenism. About one-third of patients have a monoclonal gammopathy, which may complicate distinction from lymphoplasmacytic lymphoma (LPL). In the peripheral blood, the lymphocytes may have a “villous” morphology with an eccentric nucleus and thin projections from the opposing pole of the cell. In other cases, the lymphocytes may be more “plasmacytoid.” In the bone marrow, sinusoidal infiltration is characteristically seen, with the neoplastic lymphocytes forming linear proliferations inside the marrow sinusoids. However, nodular involvement can also be seen. There are no specific immunohistochemical or molecular markers of marginal zone lymphoma; the diagnosis is made based on morphology and after exclusion of other low-grade B-cell lymphomas. NOTCH2 and KLF2 mutations may be seen in a minority of cases, resulting in disruption of the NF-κB signaling pathway.
Extranodal Marginal Zone B-Cell Lymphoma of Mucosa-Associated Lymphoid Tissue (MALT Lymphoma)
This neoplasm, usually an adult disease with a median age of about 60 years, constitutes about 8% of non-Hodgkin lymphomas. It arises in association with chronic antigenic stimulation or autoimmune disorders, such as Sjögren disease and Hashimoto thyroiditis. The prototypical MALT lymphoma arises in the stomach in association with Helicobacter pylori gastritis and in many cases can be cured with antibiotics. Other common sites are the lungs, salivary glands, eye, skin, and thyroid. Bone marrow involvement is uncommon but secondary spread to lymph nodes may be seen. In the involved tissue, in contrast to other low-grade B-cell lymphomas, the neoplastic infiltrate is heterogeneous, including small lymphocytes, plasma cells, and monocytoid B cells. The latter are small cells with generous, pale cytoplasm. Larger cells may be present as well; a predominance of large cells would be worrisome for transformation to a high-grade process. The lymphocytes may invade the epithelium and destroy it, forming a “lymphoepithelial lesion”; however, this finding is neither required nor specific for a diagnosis of MALT lymphoma.
There are no immunophenotypic findings specific to marginal zone lymphoma; in general, they tend to be negative for CD5, CD10, and Cyclin D1. Translocations involving the MALT1 gene on chromosome 18 and the BCL10 gene on chromosome 1 have been reported. The presence of genetic alterations portends a worse prognosis and a poor response to noncytotoxic forms of treatment.
Nodal Marginal Zone Lymphoma
This rare small B-cell lymphoma most commonly presents in the sixth and seventh decade. Because of the highly variable, nonspecific presentation and pathologic features, the diagnosis requires careful correlation of clinical and pathologic data. Most patients present with lymphadenopathy and a minority will also have cytopenias, elevated LDH, elevated M-protein or bone marrow involvement. Marginal zone lymphoma has variable morphology, but the most common histologic pattern is a diffuse nodal proliferation of a heterogeneous population of cells (lymphocytes, monocytoid cells, plasmacytoid cells, and plasma cells). IRTA1 has recently been shown to be a specific immunohistochemical marker for nodal marginal zone lymphoma. The translocations associated with extranodal marginal zone lymphoma are not seen in the nodal subtype, which tends to have simple chromosomal gains and losses.
Waldenström Macroglobulinemia (Lymphoplasmacytic Lymphoma)
This clonal disorder is characterized by the production of monoclonal IgM in association with bone marrow
infiltration by a proliferation of cells exhibiting a spectrum of differentiation, from small lymphocytes to plasma cells. The median age is about 65, and males slightly outnumber females. Some patients are asymptomatic, but most develop problems related to tissue infiltration of bone marrow, lymph nodes, and spleen by the malignant cells; circulating IgM; or amyloid.
infiltration by a proliferation of cells exhibiting a spectrum of differentiation, from small lymphocytes to plasma cells. The median age is about 65, and males slightly outnumber females. Some patients are asymptomatic, but most develop problems related to tissue infiltration of bone marrow, lymph nodes, and spleen by the malignant cells; circulating IgM; or amyloid.
The bone marrow may show any pattern of involvement, but most commonly it is a subtle interstitial proliferation. There are no specific immunohistochemical markers for LPL. Most LPLs are CD20+, CD5–, CD10–, which can make them challenging to distinguish from marginal zone lymphoma. The L265P mutation in MYD88 is present in 90% of cases. Rarely, MYD88 mutations other than L265P may be present. In addition, up to 35% of LPLs may have a nonsense or frameshift mutation in CXCR4 similar to those found in the warts, hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome.
Circulating IgM forms aggregates and binds water, sometimes resulting in hyperviscosity. The level of IgM does not necessarily correlate well with clinical manifestations. Ophthalmologic features include visual blurring and decreased acuity, associated with retinal hemorrhages, exudates, and dilated, tortuous veins (“sausage links” or “boxcars”). The increase in plasma volume can cause congestive heart failure. The impaired blood transit through the microvasculature can cause neurologic symptoms of dizziness, headache, deafness, confusion, nystagmus, vertigo, and ataxia. The combination of damage to vessel walls from the diminished blood flow and the interaction of monoclonal IgM with clotting factors and platelets can lead to bleeding, such as epistaxis, oral mucosal hemorrhage, and cutaneous ecchymoses. The IgM can precipitate on cooling, creating a type I cryoglobulinemia in up to 20% of patients, but less than 5% have symptoms related to it, such as cutaneous vasculitis, Raynaud phenomenon, or cold urticaria.
The monoclonal IgM also can behave as an autoantibody, and up to 20% of patients have a peripheral neuropathy from antibodies against glycoproteins in the nerves. It is usually a distal, symmetrical, chronic demyelinating process. The macroglobulins also may interact with red cell antigens at temperatures below 37°C, causing a chronic hemolytic anemia called cold-agglutinin disease. In cold agglutinin disease, the monoclonal IgM has anti-I activity and fixes complement in the cold (<37°C); the IgM then dissociates from red cells at core body temperature, leaving the red blood cells (RBCs) coated only with complement. The coated cells are rapidly cleared from the circulation. Red cell agglutination in this syndrome characteristically is as grape-like clusters of RBCs rather than as rouleaux.
Deposition of IgM in the kidneys can lead to proteinuria and renal insufficiency. Firm, flesh-colored papules and nodules can form from IgM in the dermis, and occasional patients can develop gastrointestinal problems of diarrhea, malabsorption, and bleeding from monoclonal protein present in the intestinal wall. Monoclonal light chains can form amyloid, which is clinically evident in about 2% of patients with Waldenström macroglobulinemia.
A normocytic, normochromic anemia is common and can arise from bone marrow infiltration, splenomegaly, hemolysis, hepcidin-related iron deficiency, dilution by increased plasma volume, and hemorrhage. On peripheral blood smear, rouleaux formation is common, and sometimes red cell agglutination leads to clumping of erythrocytes. Increased numbers of small lymphocytes, some resembling plasma cells (“plasmacytoid lymphocytes”) with abundant basophilic cytoplasm, are common. Plasma cells also may be visible.
Hairy Cell Leukemia
The median age of patients with this disease is approximately 55 years, and the male:female ratio is about 4:1. In most patients, the neoplastic B-lymphoid cells affect primarily the bone marrow and spleen, causing splenomegaly, monocytopenia, and neutropenia in most patients. About 50% have pancytopenia at the time of diagnosis. The disease, which accounts for about 2% of adult leukemias, predisposes to bacterial infections because of neutropenia, but also causes diminished cell-mediated immunity. The result is an increased susceptibility to nontuberculous mycobacteria, fungi, Listeria monocytogenes, Toxoplasma gondii, Pneumocystis pneumonia, and various viruses. Some patients have other rheumatologic disorders, including systemic sclerosis, polymyositis, and polyarteritis nodosa. Nearly all patients with this disease have palpable splenomegaly, often to gargantuan size, and about 40% have hepatomegaly.
In addition to cytopenia in one or more cell lines, peripheral smears in about 85% of cases reveal hairy cells—small-to medium-sized lymphoid cells that possess a round, kidney-shaped, oval or bilobed, and commonly eccentric, nucleus with ground-glass chromatin, but absent or inconspicuous nucleoli, and abundant, pale blue cytoplasm with numerous irregular, thin, surface projections resembling hairs. Because of associated fibrosis, bone marrow aspiration often results in a dry tap. Bone marrow biopsies are usually hypercellular and classically demonstrate interstitial infiltration with mononuclear cells possessing abundant cytoplasm and prominent cell borders, creating a “fried-egg” appearance. The fibrosis produces a net-like pattern affecting areas of hairy cell infiltration. Hairy cells have acid phosphatase activity that is resistant to tartrate and, in >95% of cases, a tartrate-resistant acid phosphatase (TRAP) stain is positive. Almost all cases of hairy cell leukemia (HCL) harbor the BRAF V600E mutation, which is not seen in other small B-cell lymphomas or hairy cell variant. In the rare variant form of hairy cell leukemia (HCL-v), however, the TRAP stain is usually negative, the white cell count is elevated, neutropenia and monocytopenia are absent, and the hairy cells have prominent nucleoli, similar to the cells of prolymphocytic leukemia. HCL-v is characterized by MAP2K1 mutations in ˜50% of cases, resulting in dysregulation of the mitogen-activated protein kinase signaling pathway.
MATURE T-CELL AND NK-CELL NEOPLASMS
The classification of these neoplasms depends on combining histologic, immunophenotypic, genetic, and especially, clinical features because morphologic and immunophenotypic abnormalities may be diverse within a specific disorder and yet similar among different diseases. As a group, these disorders have specific geographic foci (especially Asia), have infrequent lymph node involvement, demonstrate cell death (apoptosis) and necrosis, are accompanied by an increased incidence of the hemophagocytic syndrome, and often are associated with viral infections, especially with the EBV. In contrast to the B-cell lymphomas, classification of mature T-cell lymphomas according to cell of origin is difficult because of our limited understanding of T-cell ontogeny. We will discuss the most common T- and NK-cell neoplasms below.
T-Cell Prolymphocytic Leukemia
This disorder accounts for about 2% of adult small lymphocytic leukemias and is primarily a disease of people more than 50 years old, with most patients having impressive splenomegaly. Generalized lymph node enlargement and hepatomegaly are common. Skin nodules or diffuse papular rashes occur in about 25% of cases; pleural effusions or ascites occur in about the same number. The major laboratory finding is a very high white cell count, exceeding 200 × 109/L in about two-thirds of patients. Anemia and thrombocytopenia are also common. The diagnosis is established on peripheral blood films, where most leukocytes are prolymphocytes, with a well-defined central nucleolus and a deeply basophilic cytoplasm that often demonstrates blebs or protrusions. These cells are larger than normal small lymphocytes, but in 20% of cases of T-cell prolymphocytic leukemia (the small-cell variant), the cells are small, with more condensed nuclear chromatin and no apparent nucleolus on routine staining, but nucleoli are detectable, however, on electron microscopy.
Bone marrow aspirates show the same cells that are present on blood smears. Bone marrow biopsies show heavy infiltration that can be interstitial, nodular, diffuse, or mixed. The number of reticulin fibers is increased.
T-Cell Large Granular Lymphocytic Leukemia/Chronic NK Lymphoproliferative Disorder
The median age of patients with this disorder is about 60 years, and many have clinical and serologic evidence of rheumatoid arthritis. About 40% are asymptomatic at the time of diagnosis. The most prominent clinical feature is splenomegaly, present in about 50%; lymph node enlargement is rare. The white cell count is elevated because of an increased number of large granular lymphocytes, which usually appear normal and have round or oval, eccentric nuclei with condensed chromatin and abundant basophilic cytoplasm containing small or large purplish granules. To fulfill diagnostic criteria, these cells should exceed 2 × 109/L for at least 6 months without an alternative explanation, although occasionally patients with fewer cells may meet criteria for this diagnosis if they show other features of T-LGL. T-LGL must be differentiated from chronic NK cell lymphoproliferative disorder (CLPD-NK). Peripheral blood flow cytometry is helpful in identifying an increased CD8+, CD57+ T-cell population with a monotypic pattern of killer immunoglobulin-like receptors CD158a, CD158b, and CD158e. Polymerase chain reaction (PCR) for the T-cell receptor will show a monoclonal gene rearrangement in T-LGL but not in CLPD-NK. This lymphoproliferation of CLPD-NK is generally indolent, but the course may be complicated by severe neutropenia and/or thrombocytopenia, sometimes associated with bacterial infections or pure red cell aplasia. Bone marrow involvement is variable and often minimal, the typical pattern being intrasinusoidal. Patients with neutropenia usually have normal immature granulocytes but decreased neutrophils (maturation arrest), and patients with thrombocytopenia characteristically demonstrate adequate or increased megakaryocytes. Anemia may be accompanied by red cell aplasia or hypoplasia. Mutations in STAT3 and STAT5B are frequently seen.
Aggressive NK-Cell Leukemia
This disorder typically occurs in adolescents or young adults and is more common in Asians than in whites. Patients present acutely with fever, cytopenias, lymphadenopathy, and hepatosplenomegaly. Peripheral blood smear shows large atypical lymphocytes with fine cytoplasmic granules and irregular, hyperchromatic nuclei. The bone marrow involvement is variable and may be associated with hemophagocytosis. Most cases are associated with EBV infection.
Adult T-Cell Leukemia/Lymphoma
This disorder, caused by infection with the retrovirus human T-cell leukemia virus type 1 (HTLV-1), is endemic in Japan, the Caribbean, and parts of central Africa. Most patients have acquired the infection at an early age through breast milk or exposure to blood, and the lifetime cumulative risk of later developing leukemia or lymphoma, which occurs at a median age of about 55 years, is approximately 2%. The typical clinical features include generalized lymph node enlargement, hypercalcemia, hepatosplenomegaly, and skin lesions, which can be nodules, papules, or a diffuse scaly rash. Four clinical patterns occur. Most common is the acute variant, which includes constitutional symptoms, diffuse lymph node enlargement, skin rash, hepatosplenomegaly, hypercalcemia with or without lytic bone lesions, high numbers of circulating leukemic cells, and an elevated serum LDH. Some patients have opportunistic infections from decreased cell-mediated immunity. The lymphomatous variant consists of generalized lymph node enlargement without circulating leukemic cells. Hypercalcemia and elevated LDH may be present. The chronic variant has prominent skin lesions,
mostly an exfoliative process, but no hypercalcemia. The white cell count is elevated, with over 10% being leukemic cells. Serum LDH is slightly increased. The smoldering variant has a normal white cell count with less than 3% neoplastic cells, normal serum calcium and LDH, and no enlargement of lymph nodes, spleen, or liver. Pulmonary involvement and skin rashes may be present.
mostly an exfoliative process, but no hypercalcemia. The white cell count is elevated, with over 10% being leukemic cells. Serum LDH is slightly increased. The smoldering variant has a normal white cell count with less than 3% neoplastic cells, normal serum calcium and LDH, and no enlargement of lymph nodes, spleen, or liver. Pulmonary involvement and skin rashes may be present.
Peripheral blood smears classically show pleomorphic lymphocytes, with clumped chromatin, prominent nucleoli, and lobated nuclei (i.e., “flower cells”). A subset may appear more blast-like with fine chromatin. Bone marrow involvement is common and in most cases the cells are quite pleomorphic with prominent nucleoli. The bony structures may appear abnormal because of increased osteoclastic activity. The neoplastic lymphocytes secrete chemokines that drive bone resorption, resulting in hypercalcemia and osteolytic lesions. Adult T-cell leukemia/lymphoma is a neoplastic process arising from T-regulatory cells, which function to suppress the immune system. They express CD4 and CD25 and are generally negative for CD7. They also frequently express CCR4, which may be targeted therapeutically. This proliferation of T-regulatory cells and the consequent immunosuppression correlates with the frequent opportunistic infections seen in these patients.
Extranodal NK-/T-Cell Lymphoma, Nasal Type
This disorder, virtually always associated with EBV when it involves the nose, is more common in males than in females and is more frequent in Asia and Latin America than elsewhere. The disease typically occurs in adults and classically presents as a nasal mass. From these areas, it can disseminate to distant sites, such as the gastrointestinal tract, cervical lymph nodes, and the skin, where nodules and ulcers may form. When the disease originates outside the nasal cavity, systemic symptoms can occur.
The lymphoma cells commonly are intermingled with numerous benign cells, such as small lymphocytes, histiocytes, eosinophils, and plasma cells, making the disease often appear inflammatory rather than neoplastic. The process is destructive, typically causing ulceration and necrosis. It tends to occur around vessels, which it destroys. The tumor cells are diverse in size and appearance. They may have irregular and elongated nuclei, often undergoing mitosis, with granular chromatin. The cytoplasm is commonly pale. The peripheral blood film rarely discloses neoplastic cells. Similarly, the bone marrow rarely is involved with neoplastic cells. The cells are usually NK cell in origin and are usually positive for CD2, CD56, and cytotoxic granule proteins TIA-1 and granzyme B. A subset arises from cytotoxic T cells and would have a clonal T-cell receptor gene rearrangement detectable by PCR. Karyotype studies may show deletion of 6q or 8p.
Hepatosplenic T-Cell Lymphoma
Hepatosplenic T-cell lymphoma is a disorder of the innate immune system, arising from cytotoxic γ/δ T-cells. It classically arises in adolescents and young men, but a subset is associated with immunosuppression, especially patients exposed to azathioprine. The main clinical feature is marked hepatosplenomegaly without lymphadenopathy, usually accompanied by thrombocytopenia. The peripheral blood film rarely shows neoplastic cells, but they are usually present on the bone marrow aspirate as medium-sized lymphocytes with dispersed chromatin and mildly basophilic cytoplasm. In the liver and spleen, the neoplastic cells are medium sized and percolate through the hepatic and splenic sinuses. The marrow biopsy shows interstitial or intrasinusoidal infiltration with medium- to large-sized lymphoid cells with a rim of pale cytoplasm. Their immunophenotypic profile is variable, but classically the cells express CD56 and TIA1. Isochomosome 7q and trisomy 8 are common cytogenetic findings. Recent studies have shown mutations in STAT5B in about 40% of cases.
Mycosis Fungoides and Sézary Syndrome
This disorder occurs most commonly in adults, with a male: female ratio of about 2:1. It is a cutaneous T-cell lymphoma that begins as flat areas of skin scaling and erythema that may be asymptomatic or pruritic. At varying intervals, but typically after several years, it may progress to cause dusky red to violaceous plaques—sharply demarcated lesions that are elevated above the surrounding normal skin. Sometimes, lymph nodes are enlarged, but biopsies commonly show a reactive pattern, rather than neoplastic infiltrates. If the disease continues to advance, the next stage is the formation of cutaneous tumors. Only then does the lymphoma tend to spread to extracutaneous sites, typically, lymph nodes, spleen, liver, and lungs.
Confident pathologic diagnosis of mycosis fungoides can be very difficult, especially in the early stages, and numerous skin biopsies, sometimes taken over intervals of months to years, may be necessary before the characteristic findings are clearly present. The diagnostic abnormality consists of dermal and epidermal infiltration of small- to medium-sized T cells with irregular nuclei whose convolutions resemble the brain (cerebriform nuclei). In the dermis, infiltrates typically are present at the epidermal border and consist of small lymphocytes, eosinophils, and the neoplastic cells. Single or small numbers of the neoplastic cells may be present in the epidermis, but sometimes many aggregate there to form Pautrier abscesses. One variant of mycosis fungoides is pagetoid reticulosis, in which a chronic solitary plaque is present, and the neoplastic cells are located only in the epidermis. Another is follicular mucinosis, in which the lymphoma cells are present only in hair follicles, where they cause mucinous degeneration. Clinically, the lesions are indurated papules or plaques, which in hairy areas can cause alopecia.
Sézary syndrome is a systemic variant of mycosis fungoides characterized by erythroderma, generalized lymph node enlargement, and the presence of circulating neoplastic cells. As in mycosis fungoides, the skin shows dermal and epidermal infiltration with cerebriform lymphocytes. The peripheral
blood film has numerous small or large neoplastic cells with cerebriform nuclei containing condensed chromatin. Because these cells may appear in small numbers in both benign skin diseases and early stages of mycosis fungoides, most criteria require that their level exceed 1,000/mm3.
blood film has numerous small or large neoplastic cells with cerebriform nuclei containing condensed chromatin. Because these cells may appear in small numbers in both benign skin diseases and early stages of mycosis fungoides, most criteria require that their level exceed 1,000/mm3.
Peripheral T-Cell Lymphoma, Unspecified
This category includes about 50% of the peripheral T-cell lymphomas in Western countries. Most cases occur in adults, and the disease usually presents with nodal involvement, but disseminated disease is common, often with circulating neoplastic cells and affected extranodal sites, especially the skin. Constitutional symptoms, such as weight loss, fever, and fatigue, are frequent.
These lymphomas cause diffuse infiltration of lymph nodes with neoplastic cells that are variable, but most commonly medium- to large-sized cells with irregular, pleomorphic nuclei and prominent nucleoli. Vascular proliferation in the lymph node is common, and often a mixed inflammatory reaction is prominent, including eosinophils, plasma cells, small lymphocytes, and epithelioid histiocytes. Multinucleated cells resembling Reed-Sternberg cells and “clear cells” with very pale cytoplasm may be present. Two rare subtypes are the T-zone and lymphoepithelioid cell variants. The former has small- to medium-sized neoplastic cells in intact follicles. The latter has small cells and numerous clusters of epithelioid histiocytes.
Nodal Peripheral T-Cell Lymphoma with T-Follicular Helper Phenotype
Patients with this disorder tend to present with constitutional symptoms, rashes, polyclonal hypergammaglobulinemia, immunosuppression, and generalized lymphadenopathy. It generally occurs in older adults and has an aggressive course because of the associated immunodeficiency. Peripheral blood involvement is rare. Lymph nodes show prominent arborizing vasculature with expansion of the interfollicular areas by atypical T cells with clear cytoplasm (so-called “clear-cell immunoblasts”). Lymph node follicles may appear atrophic or may be absent. The cell of origin is the T-follicular helper cell (TFH), which classically expresses CD4 and CD10. The neoplastic T cells need to express two or three TFH-related antigens, such as PD1, CD10, BCL6, CXCL13, ICOS, SAP, and CCR5. These cells induce proliferation of follicular dendritic cells and recruit B cells to the lymph node. An IgH gene rearrangement can be seen in 30% of cases, in addition to T-cell receptor gene rearrangement, which is present in over 90%. Sequencing studies have identified frequent mutations in RHOA, IDH2, DNMT3A, and TET2 in these cases.
Anaplastic Large-Cell Lymphoma, ALK-Positive
This disorder causes about 3% of adult non-Hodgkin lymphoma and about 10% to 20% of childhood lymphomas. The usual clinical features are generalized lymph node enlargement and constitutional symptoms, including fever. The disease is commonly widespread at the time of diagnosis, typically involving such extranodal sites as skin, bone, lung, and liver. The neoplastic cells are pleomorphic, but despite this lymphoma having three variants—common, lymphohistiocytic, and small-cell—all cases include some characteristic cells called “hallmark cells” because they are present in all types. These have eccentric nuclei shaped like horseshoes or kidneys, sometimes with a perinuclear eosinophilic area. They are usually, but not always, large. In the common variant, which accounts for about 70% of cases, large hallmark cells typically predominate, and they possess abundant cytoplasm that is clear, basophilic, or eosinophilic. Multiple nuclei may occur, the chromatin usually is dispersed, and nucleoli are prominent. Sometimes, the hallmark cells are less numerous than the large neoplastic cells with rounded nuclei. The neoplastic cells are positive for CD30 and ALK. ALK is a tyrosine kinase receptor that is normally not expressed in lymphocytes. Various genetic alterations cause a fusion of the ALK gene with other partners, leading to overexpression of the ALK protein. Although molecular methods can be used to detect ALK translocations, because of the wide variety of potential partners, immunohistochemical stains are the most sensitive method of detecting ALK alterations. ALK-positive anaplastic large-cell lymphoma (ALCL) has a better prognosis than its ALK-negative counterpart (discussed below), with a 5-year survival rate of 80%. Neoplastic cells are rarely visible in the peripheral blood film, where they are large and pleomorphic. On bone marrow aspirates, they are also usually sparse. Bone marrow biopsies may show the large, pleomorphic, and sometimes multinucleated lymphoma cells in an interstitial, focal, or diffuse pattern.
Anaplastic Large-Cell Lymphoma, ALK-Negative
ALK-negative ALCL is morphologically similar to ALK-positive cases but has a different epidemiology, prognosis, and molecular background. ALK-negative ALCL tends to affect individuals in their 40s to 60s and there is a mild male predominance. In comparison with ALK-positive cases, the prognosis is poor with a 5-year overall survival of 49%. Approximately 30% of ALK-negative cases harbor a DUSP22-IRF4 rearrangement, and these cases have a prognosis comparable with ALK-positive ALCL. A smaller subset of patients (<10%) have translocations involving TP63, and these cases have extremely poor outcomes (17% 5-year overall survival). These molecular alterations can be detected by FISH or next-generation sequencing platforms.
HODGKIN LYMPHOMA
Hodgkin lymphomas constitute about 30% of all lymphomas. Hodgkin lymphomas are defined by the presence of diagnostic tumor cells, known as Reed-Sternberg cells, in the appropriate cellular environment. The WHO classification divides this disorder into two major categories:
nodular lymphocyte predominant Hodgkin lymphoma and classical Hodgkin lymphoma, which has four subtypes—nodular sclerosis, mixed cellularity, lymphocyte-rich, and lymphocyte-depleted Hodgkin lymphoma. In classical Hodgkin lymphoma, the Reed-Sternberg cells are single or multinucleated cells with prominent central nucleoli. They are derived from a pre-apoptotic germinal center B cell in which there is epigenetic silencing of B-cell genes and downregulation of B-cell transcription factors Oct2 and BOB.1. These cells coexpress surface antigens CD15 and CD30 with weak nuclear expression of PAX-5, a B-cell marker. They are negative for CD45. In contrast, the neoplastic cell population in nodular lymphocyte predominant Hodgkin lymphoma is the “LP” or “popcorn” cell, which is derived from a germinal center B cell with somatic hypermutation. These cells are further along the B-cell development pathway and consequently express B-cell markers like CD20, Oct2, and BOB.1. They are negative for CD15 and CD30.
nodular lymphocyte predominant Hodgkin lymphoma and classical Hodgkin lymphoma, which has four subtypes—nodular sclerosis, mixed cellularity, lymphocyte-rich, and lymphocyte-depleted Hodgkin lymphoma. In classical Hodgkin lymphoma, the Reed-Sternberg cells are single or multinucleated cells with prominent central nucleoli. They are derived from a pre-apoptotic germinal center B cell in which there is epigenetic silencing of B-cell genes and downregulation of B-cell transcription factors Oct2 and BOB.1. These cells coexpress surface antigens CD15 and CD30 with weak nuclear expression of PAX-5, a B-cell marker. They are negative for CD45. In contrast, the neoplastic cell population in nodular lymphocyte predominant Hodgkin lymphoma is the “LP” or “popcorn” cell, which is derived from a germinal center B cell with somatic hypermutation. These cells are further along the B-cell development pathway and consequently express B-cell markers like CD20, Oct2, and BOB.1. They are negative for CD15 and CD30.
The neoplastic cells rarely appear in the peripheral blood smear. Even with marrow involvement, they are also uncommon in the bone marrow aspirate, where they appear as large cells with two nuclei and prominent nucleoli. On bone marrow biopsy, neoplastic cells are present in about 10% of cases and usually exist in a mixture of small lymphocytes, eosinophils, and macrophages. The pattern can be either focal or diffuse. Variations include the presence of numerous Reed-Sternberg cells with few other cells, a fibrotic marrow with few neoplastic cells, and a hypocellular marrow with scattered foci of neoplastic and reactive cells.
Nodular Lymphocyte Predominant Hodgkin Lymphoma
This group accounts for 5% of Hodgkin lymphoma and typically occurs in the fourth to sixth decade of life. It usually causes localized lymph node enlargement in cervical, axillary, and inguinal lymph nodes. The histology is characterized by a nodular infiltrate of small lymphocytes and histiocytes with scattered large atypical cells called L&H cells (“lymphocytic and/or histiocytic Reed-Sternberg cells”). In contrast to classical Hodgkin lymphoma, neutrophils and eosinophils are rare. There may be morphologic overlap between cases of nodular lymphocyte-predominant Hodgkin lymphoma and T-cell/histiocyte-rich forms of large B-cell lymphoma; a broad panel of immunohistochemical stains is necessary to resolve this differential. The disease has a fairly indolent course with a good prognosis despite frequent relapses. The overall survival is 80% at 10 years.
Classical Hodgkin Lymphoma
This group accounts for 95% of cases of Hodgkin lymphoma and has a bimodal age distribution with a peak between 20 and 30 years and a smaller peak in the elderly. Most patients present with localized lymph node enlargement affecting cervical, mediastinal, axillary, or para-aortic regions. Primary extranodal involvement is rare, as is bone marrow infiltration, except in advanced disease or severely immunodeficient individuals. All subtypes of classical Hodgkin lymphoma will have variably numerous Reed-Sternberg cells with the identical immunophenotype (described above). The epidemiology, association with immunosuppression and EBV, and the background reactive infiltrate will vary by subtype.
Nodular Sclerosis Hodgkin Lymphoma
This type accounts for about 70% of classical Hodgkin lymphoma, with a median age of 28 and equal gender distribution. Most patients present with stage II disease, and about 40% have B symptoms. The most common site of involvement is the mediastinum. Lymph nodes have a thickened capsule with broad bands of fibrosis dividing the lymphoid tissue into nodules composed of a polymorphous cell population, with small lymphocytes, plasma cells, eosinophils, and macrophages admixed with less numerous Reed-Sternberg cells. Because of formalin fixation, the cytoplasm retracts around these large cells, leaving a white halo (or “lacuna”) around them, earning them the title “lacunar cells.” These neoplasms are EBV-associated in 10% to 40% of cases.
Mixed Cellularity Hodgkin Lymphoma
This type accounts for about 25% of cases of classical Hodgkin lymphoma, is more frequent in male patients, as well as in patients with HIV or those from the developing world, and shows evidence of EBV infection in at least 70% of cases. The average age is about 35 to 40 years. Patients commonly have advanced disease and B symptoms, with widespread enlargement of peripheral lymph nodes. The normal nodal architecture is effaced by an expansion of the interfollicular areas by an infiltrate of eosinophils, histiocytes, and plasma cells admixed with numerous Reed-Sternberg cells. Fibrosis is noticeably absent.
Lymphocyte-Rich Classical Hodgkin Lymphoma
In this disease, which constitutes about 5% of classical Hodgkin disease, peripheral lymph node enlargement is the main feature, and patients usually have stage I or II disease without B symptoms. About 70% of patients are male. The lymph nodes can show a diffuse or nodular pattern with numerous small lymphocytes, no neutrophils or eosinophils, and scattered Reed-Sternberg and lacunar cells. About 40% have evidence of infection with EBV.
Lymphocyte-Depleted Classical Hodgkin Lymphoma
This type, which accounts for less than 1% of classical Hodgkin lymphoma, often is associated with HIV infection. The median age is 37 years, and about 75% of patients are male. Peripheral lymph nodes are less commonly involved than are retroperitoneal lymph nodes, abdominal organs, and bone marrow. Most patients have advanced disease with B symptoms. The lymph nodes have a relative paucity of lymphocytes and sheets of neoplastic cells. Evidence of Epstein-Barr infection is common.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


