Lymphomas in Children
Childhood lymphomas are gratifying to treat because of their curability. Central to this progress has been single and multi-institutional clinical trials that, in turn, have benefited from advances in our understanding of the normal immune system, through diagnostic imaging and pathology. The primary focus of pediatric trials in the past decade has been stratification of patients into risk groups to refine treatment as follows: (a) less morbid therapy in children with a favorable prognosis, and (b) intensification of therapy in children with an unfavorable prognosis. Furthermore, early response to therapy has been recognized as a very important tool in identifying patients with more sensitive disease in whom therapy can be reduced or patients with more aggressive disease that may benefit from therapy escalation.
Commensurate with the increasingly effective use of chemotherapy has been a more restrictive role for radiotherapy. Our recognition of the long-term sequelae of therapy has played a prominent role in the development of effective treatment strategies.
 HODGKIN LYMPHOMA
HODGKIN LYMPHOMA
Historically, irradiation techniques and doses used successfully in adults caused substantial morbidities (primarily musculoskeletal growth inhibition) in children.1,2 Contemporary treatment programs use a risk-adapted approach in which patients receive varying intensities of multiagent chemotherapy and low-dose involved field irradiation.3–6,7–9,10–11,12,13,14,15,16,17,18,19–20,21–24,25–27 It is now clear that the vast majority of children with Hodgkin lymphoma (HL) can be cured, prompting increased attention to devising nonmorbid therapy for these patients. Considering the excellent outcome for the majority of children and adolescents diagnosed with HL, the identification of biologic factors predicting very good or very poor outcome is critical to direct future refinements in therapy.
 EPIDEMIOLOGY
EPIDEMIOLOGY
Lymphomas are the third most common form of childhood cancer, comprising 15% of cancer diagnoses in individuals younger than age 20 years. Overall, pediatric HL is more common than non-Hodgkin lymphoma (NHL), with an annual incidence rate of 12.8 per 1 million children (≤19 years old).28
Childhood HL has unique epidemiologic presentations that vary geographically:
• The childhood form occurs in patients age 14 years or younger. It is rare in children <4 years of age, usually occurring in children >10 years. The childhood form of HL is associated with increasing family size and decreasing socioeconomic status. Early and intense exposure to an infectious agent has been speculated to increase the risk for the childhood form of HL.29,30
• The young adult form affects patients aged 15 to 34 years and has a roughly equal incidence between older adolescent males and females. In contrast to childhood HL, young adult HL is associated with a higher socioeconomic status, as found in high-income countries. The risk for young adult HL also decreases significantly with increased sibship size and later birth order.31 Delayed exposure to an infectious agent has been proposed as a risk factor for the development of young adult HL because its epidemiologic features are similar to those seen with paralytic poliomyelitis.30 However, Chang et al.29 demonstrated that early exposure to other children at nursery school and day care seems to decrease the risk of young adult HL, most likely by facilitating childhood exposure to common infections and promoting maturation of cellular immunity.
Biology
HL is unique among the lymphomas because the malignant Hodgkin and Reed-Sternberg (HRS) cells, lymphocytic and histiocytic (L&H) cells, and their variants account for <1% of the tumor cell population. Identical immunologic gene rearrangements in HRS and L&H cells support their origin from a single transformed B cell that subsequently undergoes monoclonal expansion.32,33,34 Two distinct immunophenotypes of HL exist. The first immunophenotype, characteristic of L&H cells, consistently expresses CD20 and J chain and does not express CD30 and CD15.34,35 The second immunophenotype, characteristic of HRS cells, consistently expresses CD30, frequently expresses CD15, and does not express J chain. These immunophenotypes differentiate lymphocyte-predominant HL from classical HL, as outlined in the World Health Organization’s (WHO) classification.36,37 Substantial data exist to support a strong association between HL and the Epstein-Barr virus (EBV).38,39 The incidence of EBV-associated HL varies by age, gender, ethnicity, histologic subtype, and regional economic level.39 EBV-positive tumor genomes are more frequent in children <10 years of age and in those who live in low-income countries (see Chapter 77). HL is also associated with congenital (e.g., ataxia telangiectasia) and acquired (e.g., human immunodeficiency virus [HIV]) immunodeficiency states.40 Familial cases of HL suggest a genetic predisposition to the disease or a common environmental exposure. Concordance of HL has been observed in first-degree relatives (particularly of the same gender) and in parent–child pairs.41
Pathologic Classification
The WHO classification categorizes HL into two major groups of HL—classical HL and lymphocyte predominant (LP) HL—according to their biological and clinical features.36,37 Classical HL is further subclassified into nodular sclerosing (NS), mixed cellularity (MC), lymphocyte-rich (LR), and lymphocyte-depleted (LD) histologies based on their unique morphology. The pathologic characteristics are the same as in adults and are described in Chapter 77. The relative distribution of the subtypes differs in younger children compared with adolescents and adults. LP is relatively more common (13%) in children younger than age 10, whereas LD is exceedingly rare. Although NS is the most common subtype in all age groups, it is more frequent in adolescents (77%) and adults (72%) than in younger children (44%). Conversely, MC is more common in younger children (33%) than in adolescents (11%) or adults (17%).42
Clinical Presentation
Most children (80%) present with cervical lymphadenopathy. The lymph nodes are often fixed, firm, rubbery, and painless. Mediastinal involvement is present in 76% of adolescents but only in 33% of children aged 1 to 10 years.43 Occasionally patients are diagnosed after the onset of respiratory distress, although isolated mediastinal disease is rare as is isolated infradiaphragmatic HL, both occurring in <5% of patients. One-third of patients have one or more of the so-called B symptoms at diagnosis (unexplained fever >38°C with recurrent episodes during the previous month, drenching night sweats recurrent during the previous month, or weight loss of more than 10% in the 6 months preceding diagnosis).43,44 Cytokine production induced by the HRS cells are responsible for these manifestations as well as a variety of other clinical and pathologic features of HL, including anorexia, pruritus, fibrosis, eosinophilia, thrombocytosis, plasmacytosis, and immunodeficiency.45
Diagnostic Work-Up
The diagnosis of HL is made by lymph node biopsy and is confirmed pathologically by the presence of HRS cells and their mononuclear variants. The diagnosis is facilitated through an excisional lymph node biopsy, which enables evaluation of the malignant HRS cells within the characteristic architectural changes associated with the specific histologic subtypes. The recommended procedures for pretreatment evaluation of the child with HL are similar to those for the adult. Because bone marrow involvement at initial presentation is uncommon and rarely occurs as an isolated site of extranodal disease, bone marrow biopsy can be restricted to patients with B symptoms or stage III or IV disease.
Imaging studies of the thorax include a chest radiograph and a computed tomography (CT) scan, which alters treatment decisions in at least 10% of patients through delineation of radiographically inapparent disease involving subcarinal, hilar, or cardiophrenic angle nodes, and in extranodal sites (pleura, chest wall, or pericardium). Using the ratio of the measurement of the mediastinal mass to the maximum diameter of the intrathoracic cavity on an upright chest radiograph is a standard method to assess mediastinal bulk (mediastinal to thoracic ratio of ≥33%). Infradiaphragmatic disease is best assessed by CT scan or magnetic resonance imaging (MRI). The optimal CT evaluation requires oral and intravenous contrast agents to distinguish lymphadenopathy from other infradiaphragmatic structures. CT evaluation of abdominopelvic disease may be compromised in suboptimally contrasted studies and in children who lack retroperitoneal fat. In these cases, MRI may provide better assessment of disease involvement in the retroperitoneal lymph nodes.46 Nuclear imaging studies such as positron emission tomography (PET) scanning are helpful in staging and monitoring treatment response, particularly in cases with persistent radiographic abnormalities or “rebound” thymic growth after completion of therapy.47 Splenic and hepatic involvement by HL is suggested by the presence of enlarged organs with areas of abnormal density on CT or MRI scans, as well as increased fluorodeoxyglucose (FDG) uptake on PET scan.
Staging Systems
As for adults, children with HL are staged according to the system devised at the Ann Arbor Staging Conference in 1970 and revised at the Cotswolds meeting. Refer to Chapter 77 for complete staging information.
Prognostic Factors
As the treatment of HL has improved, the factors that influence outcome have diminished in importance. However, several factors continue to influence the choice and success of therapy. These factors are interrelated in that disease stage, bulk, and biologic aggressiveness are frequently codependent.48,49 A further complication in the determination of prognostic factors is that relevant variables often depend on staging evaluation and treatment. Similarly, patients with early stage disease have different prognosticators than patients with advanced stage disease.48 Illustrating the complexity of this subject are data from a multi-institutional study that specifically address prognostic factors in 320 children with clinical stage I to IV disease. On univariate analysis stage IV, NS HL, B symptoms, white blood cell count (WBC) of ≥11,500/mm3, hemoglobin ≤11.0 g/dL, bulky mediastinal disease, extranodal disease, and erythrocyte sedimentation rate (ESR) ≥50 mm per hour were significant for inferior disease-free survival (DFS) and overall survival (OS). By multivariate analysis, male gender; stage IIB, IIIB, or IV disease; WBC ≥11,500/mm3; and hemoglobin ≤11.0 g/dL were significant for inferior DFS and OS. Prognosis was associated with the number of adverse factors.50
The following are some generalizations regarding prognostic variables derived from both adult and pediatric data:
• Stage of disease is the most significant prognosticator of treatment outcome. Stage IV disease with multiple organ involvement confers an exceptionally poor prognosis when managed with conventional therapy.50,51
• Bulk of disease is reflected in the disease stage but more specifically is determined by the volume of distinct areas of involvement and the number of disease sites. Large mediastinal adenopathy, defined as a mass of more than one-third of the intrathoracic diameter, is associated with an increased risk of disease recurrence, particularly when managed with radiation therapy alone.43,52–55 However, overall survival remains high because of the effectiveness of salvage chemotherapy.52,53,55 Patients with multiple sites of involvement (usually defined as three or more sites) have an inferior freedom from relapse and survival in some but not all reports.54–57
• Presence of systemic symptoms (B disease), which result from cytokine secretion, reflects biologic aggressiveness and correlates with an increased risk of relapse compared with the absence of such symptoms (A disease).44,50
• Abnormally high levels of certain serum markers have been reported to be prognosticators of a negative outcome.58 Whether these elevated levels are caused by more malignant biology of disease or by increased tumor volume is unclear. In most reports, independent prognostic significance for serum markers is unproven except in select clinical scenarios.48,50
• Histologic subtype may correlate with prognosis. LD histology confers a worse outcome than do the other subtypes, but it is exceedingly rare.55 Patients with LP histology are more commonly early stage59 and have an excellent outcome. Most modern combined modality trials do not show any histology survival difference.21,50 Recently, the Children’s Oncology Group (COG) reported a survival advantage for patients with mixed cellularity HL treated in their low-risk study AHOD0431.60
• Finally, patient age appears to be a determinant of patient outcome.42,61 Children <10 years of age have been observed to fare better than older patients, possibly because they are more likely to have early stage or A disease.42,62
The use of these prognostic factors is reflected in the risk grouping that various pediatric trials employ for assignment of therapy. Such stratification is discussed below in the “Combination Chemotherapy, Combined Modality Therapy, and Risk-Adapted Therapy” section and is reflected in the summary treatment recommendations.
General Management
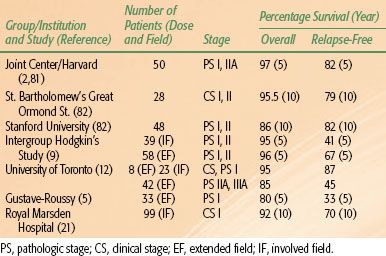
Although HL is one of the few pediatric malignancies that has an adult counterpart with similar natural history and biology, determination of the optimal therapeutic approach for children with this disease is complicated by their longevity and increased risk for adverse treatment-related side effects over time. In particular, radiation therapy doses and fields used in adults can produce significant musculoskeletal growth retardation and cardiovascular and pulmonary effects, as described later in this chapter.1,2,63 Consequently, the various successful approaches to treatment of HL in children (Tables 89.1, 89.2, and 89.3) must be considered in terms of efficacy and morbidity, and this is influenced by the developmental status of the patient.
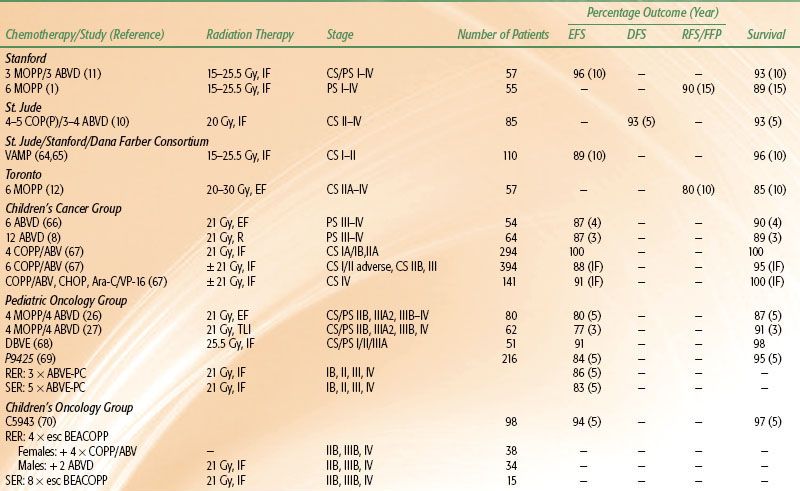
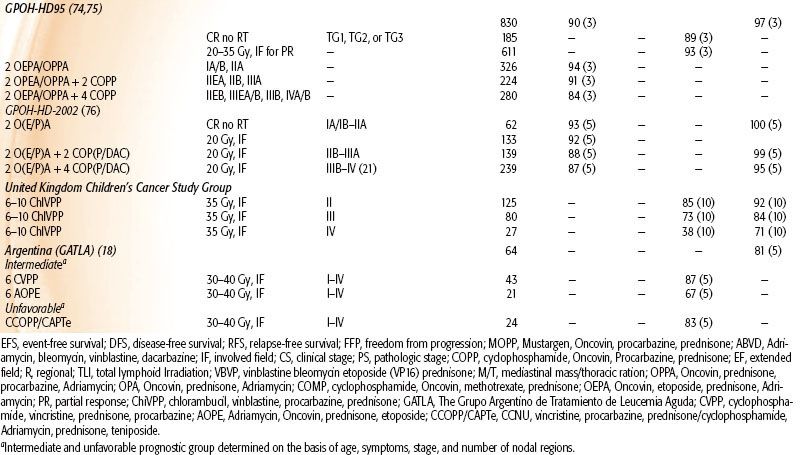
TABLE 89.1 TREATMENT RESULTS OF COMBINED-MODALITY TRIALS OF PEDIATRIC HODGKIN LYMPHOMA

Radiation Therapy
Radiation therapy (RT) to extended fields to a curative dose range of 30 to 40 Gy produced the first cures in patients with HL, and similar treatment paradigms were used for adults and children. Five-year DFS rates following extended-field radiation therapy in surgically staged children with early stage disease ranged from 60% to 80%.4,5,9,64,65 However, it became apparent that the late effects of RT posed significant risks in terms of morbidity and mortality in pediatric HL survivors.1,52,63,83–88,89,90–92
Combined-modality treatment programs evolved in an effort to reduce therapy-related toxicities; reduced radiation dose was combined with non–cross-resistant chemotherapy in pediatric patients. These regimens reduced the dose-related toxicity of both chemotherapeutic agents and RT and improved DFS in advanced stage patients.1,8,10,11,26,27,93 Advances in radiation technology and diagnostic imaging led to a reduction in RT field size, permitting involved-field radiotherapy (IFRT) treatment approaches that more effectively shield normal tissues. Although radiation alone has been largely abandoned as a treatment strategy in children, risk-adapted treatment strategies still integrate low-dose (15 to 25.5 Gy) IFRT as a critical component of combined-modality regimens and as salvage therapy for patients with refractory or relapsed disease.
TABLE 89.2 TREATMENT RESULTS OF CHEMOTHERAPY ALONE IN PEDIATRIC HODGKIN LYMPHOMA TRIALS
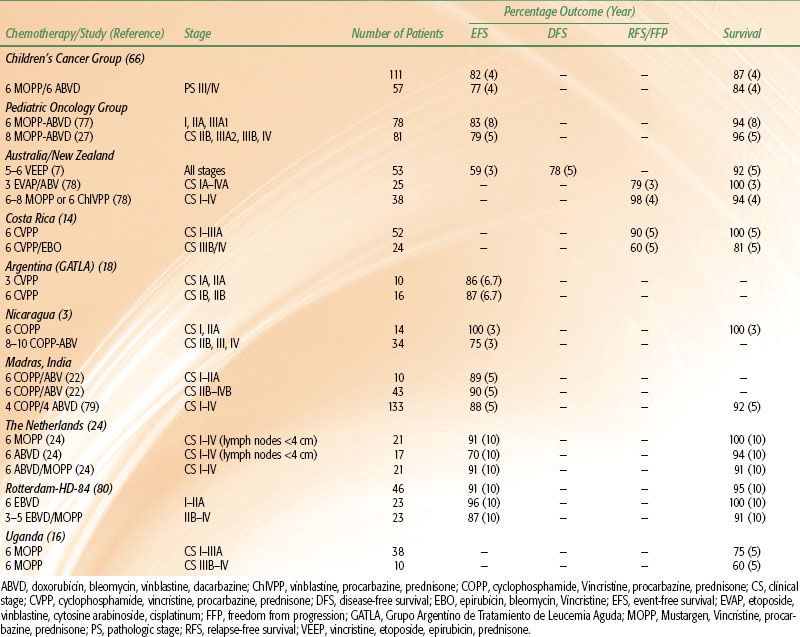
TABLE 89.3 TREATMENT RESULTS IN CHILDREN WITH HODGKIN LYMPHOMA WITH RADIATION THERAPY ALONE
Combination Chemotherapy, Combined Modality Therapy, and Risk-Adapted Therapy
The development of non–cross-resistant chemotherapy combinations provided the first effective therapy for advanced HL and formed the cornerstone for contemporary risk-adapted therapy. The specific chemotherapeutic combinations have changed as their morbidities have become better understood, but most treatment regimens include MOPP (mechlorethamine, vincristine, procarbazine, and prednisone), ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine), or therapies derived from one of the combinations (Table 89.2).
Despite the excellent disease control with ABVD,6,85,93,94 bleomycin and doxorubicin cause pulmonary and cardiovascular damage, respectively. To reduce the treatment-related toxicities associated with 6 cycles of either MOPP or ABVD, strategies alternating the two regimens, and using fewer cycles of each, have been developed. The combined ABVD and MOPP regimens used for children have produced excellent disease control with apparent diminished toxicity.10,11,27
Contemporary therapy for pediatric HL using chemotherapy alone or combined-modality therapy produces long-term DFS in 85% to 100% of patients with localized disease and 70% to 90% of patients with advanced disease.3,6,7–8,10,11,13,14,15,16, 17,18,20,22–24,25–27,67,71,73,74,77,78,83,94,95 Single-modality chemotherapy treatment usually involves more cycles of chemotherapy, whereas combined-modality therapies prescribe low-dose, involved-field radiation in lieu of several chemotherapy cycles. Although early treatment results appear comparable with results of more mature trials of combined-modality therapy, the long-term efficacy and treatment toxicities associated with chemotherapy alone have not been reported. Earlier randomized pediatric trials prospectively comparing outcomes in patients treated with chemotherapy alone with those treated with combined-modality therapy have not definitively established the superiority of one treatment approach over the other.26,93 However, other trials suggest a better outcome in patients with intermediate or advanced disease treated with combined-modality therapy.26,74 Children’s Cancer Group (CCG) investigators closed enrollment into a randomized controlled trial comparing outcomes in patients treated with contemporary risk-adapted combined-modality therapy with COPP/ABV (cyclophosphamide, vincristine, prednisone, procarbazine/doxorubicin, bleomycin, vinblastine) hybrid chemotherapy and low-dose, involved-field radiation to those treated with COPP/ABV chemotherapy alone based on a significantly higher 3-year event-free survival (EFS) in patients randomized to also receive radiotherapy.67 However, early follow-up did not demonstrate a significant difference in OS among the groups due to the successful retrieval of relapsed patients following salvage therapy. Similarly, the German HL-DAL-90 protocol treated intermediate- and high-risk patients with 2 cycles of OPPA (vincristine, prednisone, procarbazine, doxorubicin) or OEPA (vincristine, etoposide, prednisone, doxorubicin) (females and males, respectively), followed by 2 to 4 cycles of COPP. All patients received 20 to 35 Gy IFRT. Five-year EFS in the intermediate- and high-risk groups was 93% and 86%, respectively, which was comparable to that seen in the low-risk group. OS (5-year) for all three risk groups was ≥94%.17,20,74,75 In the subsequent HL-95 trial, which employed the same chemotherapy but omitted IFRT in complete responders, the EFS for intermediate- and high-risk patients combined who did not receive radiotherapy dropped to 79% compared with 91% for those who did get radiotherapy.74
Currently, consensus regarding the optimal therapy for children and adolescents with HL has not been established. Treatment with chemotherapy alone is preferred by many investigators desiring to avoid long-term sequelae of radiation, including musculoskeletal growth impairment, cardiovascular dysfunction, and solid tumor carcinogenesis. However, single modality chemotherapy protocols typically use higher cumulative doses of chemotherapeutic agents with dose-related toxicity, especially alkylating agents. Moreover, low-dose involved-field radiation therapy has substantially reduced many of the undesirable radiation-associated sequelae. Therefore, current investigations aim to selectively combine chemotherapy and radiation in an effort to improve the therapeutic ratio.
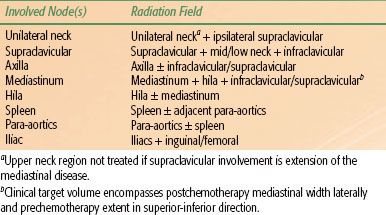
Ongoing risk-adapted trials aim to identify patients who require radiation to optimize DFS and patients who can be cured by limited chemotherapy alone (Table 89.4).
TABLE 89.4 INVOLVED FIELD RADIATION GUIDELINES
Favorable Risk
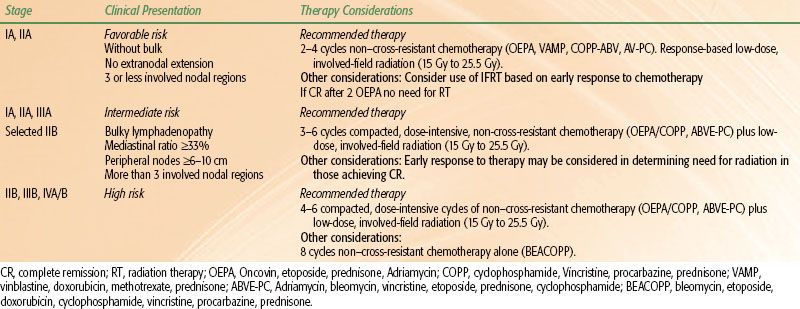
In the 1990s, pediatric HL investigators evaluated risk-adapted therapies using fewer cycles of combination chemotherapy and lower radiation doses and treatment volumes in clinically staged patients with favorable disease presentations. Favorable disease presentations were characterized by localized nodal involvement in the absence of B symptoms and bulky mediastinal lymphadenopathy. The number of involved nodal regions, the presence of extranodal extension, hilar lymphadenopathy, and peripheral nodal bulk comprised the other risk factors considered in some of these studies. Most treatment regimens prescribed 2 to 4 cycles of novel chemotherapy combinations that limited or omitted alkylating agent and anthracycline chemotherapy and bleomycin. The results of these trials demonstrated that excellent treatment outcomes could be maintained in patients with favorable presentations of localized HL using minimal therapy.13,17,19,20,25,65,67,68,71,74,96 Ongoing trials are testing the feasibility of omitting chemotherapy in patients with excellent response to chemotherapy.
Intermediate and Unfavorable Risks
In most clinical trials, intermediate risk consists of stage I, II, and IIIA patients with unfavorable disease features such as more than three nodal sites, presence of bulky mediastinal lymphadenopathy (mediastinal ratio ≥33%, peripheral nodal mass ≥6 to 10 cm), or extranodal extension. High-risk HL patients are those with stage IIIB and any with stage IV. Patients with stage IIB disease are sometimes considered intermediate and sometimes high risk according to the study group. Two treatment approaches are used for intermediate and high-risk patients, both of which include chemotherapy regimens derived from the original MOPP and ABVD combinations. MOPP has largely been replaced by COPP because cyclophosphamide is less myelosuppressive and leukemogenic than mechlorethamine.97
In the conventional treatment approach, combination chemotherapy is administered on a twice monthly schedule for 6 to 8 months.8,10,11,17,19,20,26,27,67,72,74,93 More recent trials are evaluating the second treatment approach, which has been used successfully in adults with HL. These protocols prescribe dose-intensive, multiagent chemotherapy in an abbreviated schedule over a period of 3 to 5 months. Prototypes of the dose-intensive therapy include the MOPP/ABV hybrid, Stanford V, or BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone) regimens.67,70,98,99,100 These treatments alternate myelosuppressive and nonmyelosuppressive chemotherapy combinations in a weekly schedule with the aim of reducing the development of resistant disease. Growth factor support is required to facilitate bone marrow recovery and maintain the compacted treatment schedule. Low-dose, involved-field radiation therapy generally is used to consolidate remission after completion of chemotherapy. In ongoing pediatric trials, the response to chemotherapy influences the ultimate amount of therapy. For example, in the Stanford/St. Jude/Dana Farber protocols, rapid early responders to chemotherapy, defined by complete response after 2 cycles of chemotherapy, are being targeted for reduced treatment (less or no radiation therapy, depending on the risk group). In the last COG trial for high-risk patients, rapidity of response was evaluated after 4 cycles of BEACOPP. Rapid responders received consolidation therapy according to gender in order to avoid or minimize sex-specific long-term toxicities of therapy. Females received 4 cycles of COPP/ABV without IFRT to avoid secondary breast cancers, while males received 2 cycles of ABVD with IFRT. Slow responders received 4 more cycles of BEACOPP followed by IFRT. OS was 97%, with significant acute hematologic toxicity and two patients developing secondary leukemia.70 The current European trial, based on the long-standing German experience, is also testing a gender-based approach in which a procarbazine-free OEPA/COPDAC (cyclophosphamide, vincristine, prednisone, dacarbazine) is given to boys and OEPA/COPP to girls, in order to reduce the alkylator exposure in boys and maintain fertility.76
The last COG intermediate risk HL study treated patients with stage IA or IIA bulky, IB, IIB, IIIA, or IVA disease with 2 cycles of ABVE-PC (doxorubicin, bleomycin, vincristine, etoposide, prednisone, cyclophosphamide) prior to response evaluation. Those with a rapid early response to therapy (>60% reduction in the tumor dimension) and those who achieve complete recovery after an additional 2 cycles of the same chemotherapy were randomized to 21 Gy IFRT or no additional treatment. Patients with slow early response after 2 cycles of chemotherapy were randomized to either standard therapy (an additional 2 cycles of ABVE-PC + 21 Gy IFRT) or intensified therapy (2 cycles of ABVE-PC, 2 cycles of DECA [dexamethasone, etoposide, cisplatin, cytarabine] + 21 Gy IFRT). Early results show an improved outcome for rapid early responders of 87% compared with 78% for slow early responders (P = .0001), while radiotherapy did not improve outcome in the rapid early responders (P = .07).101
Sequence of Therapy
The most effective sequence of therapy in the setting of combined chemotherapy and irradiation is not unequivocally established. However, chemotherapy is usually the first modality. This allows assessment of drug response, maximization of the amount of drug treatment, and shrinkage of disease and more limited fields of irradiation. Rarely, focal irradiation prior to chemotherapy is necessary due to significant symptomatic compromise such as airway obstruction.
Refractory or Relapsed Disease
Treatment failures in pediatric HL patients typically develop within the first 3 years, although late relapses have been reported, particularly in patients with lymphocyte-predominant HL. The most common site of relapse following risk-adapted therapies remains the primary site of disease.102 Because of the excellent outcome for the majority of children and adolescents with HL, investigations regarding salvage strategy have been limited. Prognosis following relapse is dependent on the primary therapy given and the timing of relapse. Standard multiagent chemotherapy and radiation therapy may salvage 40% to 50% of patients who relapse 1 or more years after primary therapy, but treatment complications such as second malignancies may reduce long-term survival. More aggressive salvage chemotherapy without stem cell transplant has been shown to provide satisfactory outcome for patients with late relapses.103 Patients with primary refractory disease (i.e., those unresponsive to initial therapy or who relapse within 1 year of primary therapy) have a very poor prognosis. Intensive cytoreductive chemotherapy followed by myeloablation and autologous hematopoietic stem cell transplantation is reported to salvage 30% to 60% of these patients, but relapses after 5 years have been observed, and long-term treatment complications may predispose to early mortality.104,105–107
Historically, because of higher treatment-related mortality, allogeneic transplantation did not provide a survival advantage compared with autologous stem cell transplantation. However, allogeneic bone marrow transplantation has been shown to be a useful treatment in healthy patients who have failed or cannot have an autologous transplant.108
TABLE 89.5 GUIDELINES FOR TREATMENT SELECTION IN PEDIATRIC HODGKIN LYMPHOMA
Techniques of Radiation Therapy
As discussed above, most children with HL will be treated with combined chemotherapy and low-dose IFRT using a risk-adapted approach. The use of radiation therapy alone has been largely abandoned due to concerns for late effects, including cardiac complications and secondary malignancies. The Patterns of Care studies demonstrated the relation between radiation technique and outcome.57 The results of patients treated with RT alone were improved in institutions that treated a large number of HL patients. The HD-DAL 90 trial (German-Austrian pediatric multicenter trial) showed that upfront centralized review of patients entered into the study altered the treatment approach for a large number of children.109 Because technique is discussed in Chapter 77, only select points applicable to children will be discussed here.
Using risk-adapted strategies, most children with HL are treated with combined chemotherapy and low-dose IFRT. The definition of involved fields depends on the anatomy of the region in terms of lymph node distribution and patterns of disease extension into regional areas. Involved fields typically should include not just the identifiably abnormal lymph nodes but also the entire lymph node region containing the involved nodes (Table 89.5). Historically, consideration has been given to radiating the treatment area bilaterally (e.g., both sides of the neck) in young children in an effort to avoid growth asymmetry. However, current combined modality treatment strategies use low radiation doses of 15 to 25 Gy and, therefore, unilateral radiation fields are appropriate if the disease is confined to one side of the neck. However, field definitions often are protocol specific.
Strong consideration must be given to excluding normal tissues in an effort to reduce the risk of acute and late effects, including secondary malignancies and cardiac complications. Every effort should be made to exclude breast tissue, and potentially the thyroid gland, in patients with isolated mediastinal disease with no evidence of axillary or neck involvement. Clearly, careful planning and judgment are necessary. With the use of CT-based treatment planning, normal structures can be identified and doses minimized.89,110,111–115 The treatment of involved supradiaphragmatic fields or a mantle field can pose a clinical challenge to treat precisely the lymph node regions that are involved while still protecting critical adjacent normal tissues. These fields can be simulated with the arms up over the head or down with hands on the hips. The former pulls the axillary lymph nodes away from the lungs, allowing greater lung shielding. However, the axillary lymph nodes then move into the vicinity of the humeral heads, which should be blocked in growing children. Thus, the position chosen involves weighing concerns regarding lymph nodes, lung, and humeral heads. Attempts should be made to exclude or position breast tissue under the lung–axillary blocking.
In risk-adapted strategies using combined modality therapy, radiation dose is dependent on the choice of systemic therapy that is variously defined and often protocol specific. In general, doses of 15 to 25 Gy are used, with modifications based on patient age, treatment response, the presence of bulk or residual (postchemotherapy) disease, and normal tissue concerns. In some situations, a boost of 5 Gy is appropriate. Although IFRT remains the standard when patients are treated with combined-modality therapy, response-adapted RT, which is limited to areas of initial bulk disease (generally defined as ≥5 or 6 cm at the time of disease presentation) or postchemotherapy residual disease (generally defined as ≥2 cm, or residual PET avidity), is under investigation.
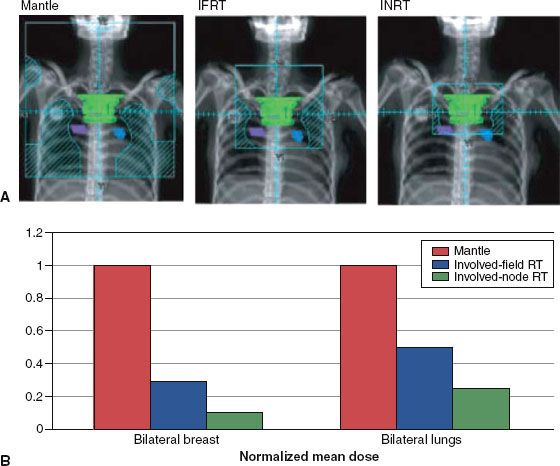
An approach studied in Europe by the European Organisation for Research and Treatment Center–Groupe d’Etudes des Lymphomes de l’Adulte is the use of involved node RT (INRT), which restricts RT to the initially involved lymph node.116 This is based on the premise that chemotherapy can effectively eliminate microscopic disease that may exist within adjacent but clinically uninvolved nodes. This concept is supported by the observation that among patients treated with chemotherapy alone, initially involved lymph nodes are the most common site of recurrence.117 INRT uses all available clinical information including pre- and postchemotherapy imaging with CT and FDG-PET scan to define the treatment field according to the prechemotherapy extent of disease.118,119 However, uninvolved lymph node regions are not necessarily included within the clinical target volume (CTV). For example, in contrast to conventional IFRT, uninvolved hila are not included in the CTV for mediastinal presentations, and the length of the treated volume is not routinely extended beyond 1 cm for the planning target volume (Fig. 89.1). However, the margins for INRT are defined differently depending on the protocol and cooperative group.112,120,121 Regardless, successive reduction in treatment volume may reduce the toxicity of therapy.
FIGURE 89.1. A: Digitally reconstructed radiographs demonstrating typical radiation therapy (RT) fields for historic mantle RT, contemporary involved-field RT (IFRT), and involved node RT (INRT) for a female patient with stage I disease involving the upper mediastinum. The postchemotherapy volume of initially involved mediastinal nodes is shown in green. Hila are shown in blue and violet. B: Reduction in dose to breast and lung for the same patient. For each volume the prescribed dose to the CTV is the same. The resulting mean dose to breast and lung tissue with mantle RT is a set value equal to 1. The proportional reduction in normal tissue dose occurs as a result of the reduction in treated volume with IFRT and INRT. (From Hodgson DC, Hudson MM, Constine LS. Pediatric Hodgkin lymphoma: maximizing efficacy and minimizing toxicity. Semin Radiat Oncol 2007;17:230–242; copyright 2007, with permission from Elsevier.)