MOST FREQUENTLY ACQUIRED MOLECULAR ABNORMALITIES IN LUNG CANCER
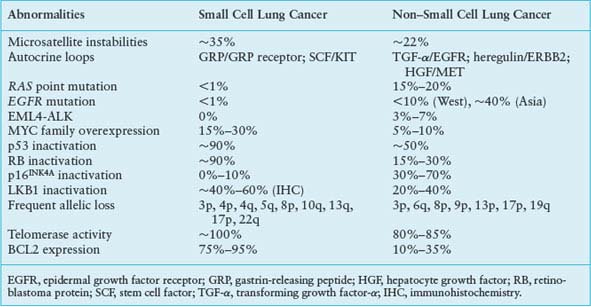
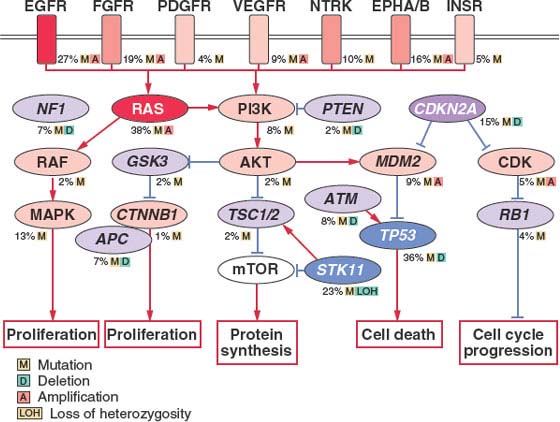
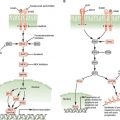
FIGURE 15.1 Significantly mutated pathways in lung adenocarcinomas. Genetic alterations in lung adenocarcinoma frequently occur in genes of the mitogen-activated protein kinase (MAPK) signaling, p53 signaling, Wnt signaling, cell cycle, and mammalian target of rapamycin (mTOR) pathways. Oncoproteins are indicated in pink to red and tumor suppressor proteins are shown in light to dark blue. The darkness of the colors is positively correlated to the percentage of tumors with genetic alterations. Frequency of genetic alterations for each of these pathway members in 188 tumors is indicated. (From Macmillan Publishers Ltd, ref. 113, copyright 2007, with permission.).
Researchers are optimistic that molecular epidemiology will help identify individuals at the highest risk of developing lung cancer. Such information, in addition to the smoking history, will be of great value in new lung cancer screening trials and in chemoprevention trials to identify persons at highest risk of developing lung cancer.
MOLECULAR CHANGES IN PRENEOPLASIA
Before lung cancer is clinically recognizable, a series of morphologically distinct changes (hyperplasia, metaplasia, dysplasia, and carcinoma in situ) are thought to occur. Whether one cell of origin leads to all histological variants is unclear. It is believed that dysplasia and carcinoma in situ represent true preneoplastic changes. These sequential changes found within squamous cell cancers that arise from central bronchi have long been recognized. Although the exact cell of origin for lung cancer is unknown, it is thought that type II epithelial cells have the capacity to give rise to lung adenocarcinomas, while cells of neuroendocrine origin are likely precursors of SCLC.
It is evident that preneoplastic cells contain several genetic abnormalities identical to some of the abnormalities found in overt lung cancer cells. For squamous cell cancers, immunohistochemical analysis has confirmed abnormal expression of protooncogenes (cyclin D1) and tumor suppressor genes (TSGs) (p53).8 Allelotyping of precisely microdissected, preneoplastic foci of cells shows that 3p allele loss is currently the earliest known change, suggesting that one or more 3p TSGs may act as gatekeepers for lung cancer pathogenesis.9 This is followed by 9p, 8p, and 17p allele loss and p53 mutation. Even histologically normal bronchial epithelium has been shown to have genetic losses. Similarly, atypical alveolar hyperplasia, the potential precursor lesion of adenocarcinomas, can harbor Kristen rat sarcoma viral oncogene homolog (KRAS) mutations and allele losses of 3p, 9p, and 17p.10 Other genetic alterations, such as inactivation of LKB1, whose germline mutations cause Peutz-Jeghers syndrome, have also been implicated in the development of adenocarcinoma. These observations are consistent with the multistep model of carcinogenesis and a field cancerization process, whereby the whole tissue region is repeatedly exposed to carcinogenic damage (tobacco smoke) and is at risk for development of multiple, separate foci of neoplasia.11
Although all types of lung cancers have associated molecular abnormalities in their normal and preneoplastic lung epithelium, SCLC patients in particular appear to have multiple genetic alterations occurring in their histologically normal-appearing respiratory epithelium. Molecular changes have been found not only in the lungs of patients with lung cancer but also in the lungs of current and former smokers without lung cancer. These molecular alterations are thus important targets for use in the early detection of lung cancer and for use as surrogate biomarkers in following the efficacy of lung cancer chemoprevention. In this regard, it appears that the smoking-damaged respiratory epithelium has thousands of clonal patches, each containing clones of cells with 3p and other allele loss abnormalities.12 The challenge is to identify not only the prevalence and temporal sequence of molecular lesions in lung preneoplasia, but to determine which are rate limiting and indispensable and thus represent potential candidates for intermediate biomarker monitoring and therapeutic efforts.
GENETIC AND EPIGENETIC ALTERATIONS IN LUNG CANCERS
Genomic Instability and DNA Repair Genes
Similar to other epithelial tumors, lung cancer cells typically display chromosomal instability—both numeric abnormalities (aneuploidy) of chromosomes and structural cytogenetic abnormalities.13 Allele loss on chromosome 3p is thought to be the among the earliest genetic change occurring in both NSCLC and SCLC.14 In addition, nonreciprocal translocations and recurrent losses involving 1p, 4p, 4q, 5q, 6p 6q, 8p, 9p, 11p, 13q, 17p, 18q, and 22q may occur, representing changes in known and potential tumor suppressor genes.15 Polysomies or regions of gene amplifications also occur and often involve protooncogenes such as epidermal growth factor receptor (EGFR) and myelocytomatosis viral oncogene homolog (MYC).16,17 Simple reciprocal translocations are uncommonly observed in lung cancer, although translocations that give rise to BRD4-NUT,18 CRCT1-MAML2,19 SLC34A2–ROS,20 and EML4-ALK21 fusion proteins have been reported, and for the case of EML4-ALK this has been shown to drive the development and proliferation of tumors. Alterations in microsatellite polymorphic repeat sequences are found in 35% of SCLC and 22% of NSCLC.22 The underlying mechanism for this chromosomal instability has not yet been discovered.
The most powerful tumor surveillance mechanism is involved in DNA damage response and repair of errors in DNA replication.23 The DNA glycosylase 8-Oxo guanine (OGG1) specifically excises the oxidatively damaged mutagenic base 8-hydroxyguanine, which causes G:C→T:A transversions frequently found in lung cancer. Lung adenocarcinoma spontaneously develops in Ogg1 knockout mice and 8-hydroxyguanine accumulates in their genomes.24,25 Individuals with low OGG activity have a greatly increased risk of developing lung cancer. Polymorphisms in other DNA repair genes ERCC1, XRCC1, ERCC5/XPG, and MGMT/AGT have been correlated with reduction of polyaromatic hydrocarbon DNA adduct formation as well as with lower lung cancer risks in case-control studies. High expression of excision repair cross-complementation gene-1 (ERCC1) is associated with decreased response to platinum-based chemotherapy, but in contrast, overexpression of ERCC1 correlates with better overall prognosis in NSCLC,26 reflecting improved repair of lethal DNA damage by platinum, on the one hand, and greater DNA stability with less aggressive disease course, on the other. Similarly, ribonucleotide reductase M1 (RRM1) overexpression correlates with better de novo prognosis but resistance to gemcitabine. Assays of both ERCC1 and RRM1 are commercially available and are marketed as tools to guide cytotoxic therapy in NSCLC; treatment based on these assays has not yet been prospectively validated, but may prove important.
PROTOONCOGENES, GROWTH FACTOR SIGNALING, AND GROWTH FACTOR TARGETED THERAPIES
EGFR and KRAS are the two most commonly mutated protooncogenes in lung adenocarcinomas; these mutations appear to be mutually exclusive. EGFR is a transmembrane tyrosine kinase, which, when activated by binding with one of its ligands, members of the EGF family, stimulates cell proliferation. When mutated, EGFR tyrosine kinase is constitutively activated, resulting in uncontrolled proliferation, invasion, and metastasis. Coexpression of EGFRs and their ligands, especially transforming growth factor-α, by lung cancer cells indicates the presence of an autocrine (self-stimulatory) growth factor loop. Activating EGFR mutations are observed in approximately 10% of North American and European populations and 30% to 50% of Asian populations27–32 and are significantly more common in never-smokers (100 or less cigarettes per lifetime) or light former smokers (quit 1 year or more ago and less than ten-pack per year smoking history). The leucine to arginine substitution at position 858 (L858R) in exon 21 and short in-frame deletions in exon 19 are the most common mutations seen in adenocarcinomas of the lung. These mutations result in prolonged activation of the receptor and downstream signaling through phosphorylated Akt, in the absence of ligand stimulation of the extracellular domain. EGFR mutations are both prognostic for response rate to chemotherapy and survival irrespective of therapy and are predictive of response to specific inhibitors of the EGFR tyrosine kinase—gefitinib and erlotinib.31,33,34 EGFR mutations are found almost exclusively in adenocarcinomas and occur much more frequently in tumors from never-smokers, women, and in Asian populations, explaining the increased clinical response to gefitinib noted in these subpopulations before the association with EGFR mutation was discovered.31,33,34 A recent review by Rosell et al.32 suggested patients with adenocarcinoma who are never or light remote smokers, especially women and Asians, should be screened for the presence of an EGFR mutation. The IPASS (Iressa Pan-Asia Study) trial demonstrated an improved outcome with up-front treatment with gefitinib compared with chemotherapy for patients with EGFR mutation, but on the other hand, much worse outcome compared with chemotherapy in patients with wild type EGFR.35,36 Previous suggestions that EGFR gene amplification by detected by fluorescence in situ hybridization (FISH) or immunohistochemistry may correlate with response to therapy with an EGFR tyrosine kinase inhibitor28 have not been corroborated. Cetuximab, a human murine chimeric immunoglobulin G subclass-1 (IgG-1) antibody that binds to the extracellular domain of EGFR and affects ligand-induced phosphorylation and degradation has been evaluated in patients with NSCLC. Contrary to data from colon cancer studies, neither expression of nor mutations in EGFR or KRAS appear to predict response or survival to this agent.37
ERBB2 (formerly human epidermal growth factor receptor 2 [HER2]/neu) is also a member of the erbB receptor family, along with EGFR. The ERBB2 receptor is unusual in that it does not interact with the EGF ligand family; its activation depends on heterodimerization with other erbB receptors following ligand binding. ERBB2 mutations have been detected with low frequency in lung adenocarcinomas.30 Similar prevalence of gene amplification of ERBB2 can be observed.38 A meta-analysis suggested that overexpression of ERBB2 is a poor prognostic indicator for survival in NSCLC.39 Several clinical trials examining trastuzumab (Herceptin), a recombinant humanized monoclonal antibody against ERBB2, as a single agent or in combination with chemotherapy, have failed to demonstrate a survival benefit.40 A new generation of dual irreversible EGFR/ERBB2 inhibitors is under development. These agents form a covalent bond with CYS-733 in EGFR and may be effective in patients with resistance to reversible EGFR tyrosine kinase inhibitors.41
c-KIT belongs to the PDGF/c-Kit receptor tyrosine kinase family that results in activation of the Janus-associated tyrosine kinase/signal transducers and activators of transcription (JAK-STAT), phosphoinositide 3-kinase (PI3K), and mitogen-activated protein kinase (MAPK) pathways important in cell growth and differentiation. Along with its ligand, stem cell factor (SCF), it is preferentially expressed in many SCLCs and is thought to have prognostic implications.42–45 Activation of this putative autocrine loop may also mediate chemoattraction and may provide a growth advantage in tumor cells. Although the c-KIT tyrosine kinase inhibitor imatinib reduces cell proliferation and induces cell death in several preclinical SCLC models,46 it failed to demonstrate any objective response in clinical trials.47–49 Another receptor tyrosine kinase, c-MET and its ligand, hepatocyte growth factor (HGF), play an important role in fetal lung development. Coexpression of this putative loop is observed in both NSCLC and SCLC. High HGF levels have also been associated with a poor outcome in resectable NSCLC patients; furthermore, elevated expression of MET is associated with resistance to EGFR targeted therapy,50 and improvement in patient outcome has recently been reported when monoclonal antibodies inhibiting MET are combined with erlotinib.51 The insulin-like growth factor receptor 1 (IGFR1) signaling pathway plays an important role in cancer growth and progression and has been associated with resistance to therapy. IGFR1 monoclonal antibodies and small molecule inhibitors are currently in clinical testing in patients with NSCLC. A phase 3 clinical trial of figitumumab (CP-751871) was closed early due to excess toxicities, including treatment-related deaths, in the experimental arm, so the possible role of these agents in the treatment of NSCLC remains to be determined.
In addition to protooncogene products, other growth stimulatory loops are found in lung cancer. The best known is that governed by gastrin-releasing peptide and other bombesin-like peptides, together with their receptors, which participate in lung development and repair and promote SCLC growth in cell culture via an autocrine loop. Immunohistochemical studies showed that gastrin-releasing peptide is expressed in 20% to 60% of SCLC but less frequently in NSCLCs. Although this loop is a possible therapeutic target, a phase 1 clinical trial of the anti–gastrin-releasing peptide monoclonal antibody 2A11 did not result in an objective antitumor response in patients with lung cancer.52
Other signaling pathways historically linked to embryonic development likely to be relevant for lung carcinogenesis include the Sonic hedgehog (Shh) and Notch signaling pathways. Extensive activation of the hedgehog pathway has been demonstrated within the airway epithelium during repair of acute airway injury and in a subset of SCLC.53 Notch 3 has been found to be overexpressed in about 40% of lung cancers. Inhibition of this pathway in vitro has resulted in the loss of tumor phenotypes.54 Clinical trials are under way to determine whether targeting these pathways has any clinical utility.
As downstream effectors are required for intracellular transduction of incoming growth factor or receptor signals, it is not surprising that proteins important in cytoplasmic signal transduction cascades are also implicated in carcinogenesis. Mutation of KRAS is the most frequently reported alteration in the downstream EGFR signaling pathways. The RAS gene family, in particular KRAS, can be activated by point mutations at codons 12 or 13 in exon 2 in approximately 20% to 30% of NSCLCs, with 90% of the RAS mutations observed in lung adenocarcinomas and with approximately 85% of the KRAS mutations affecting codon 12.55 KRAS mutations are rarely observed in SCLCs. Transgenic mice with oncogenic Kras alleles that are activated by spontaneous recombination events in the whole animal are highly predisposed to early onset lung cancer, further supporting the hypothesis that KRAS mutation is an early oncogenic event in lung cancer.56 Interestingly, the remaining wild type Ras allele appears to function as a potential tumor suppressor, because mice susceptible to the chemical induction of lung tumors frequently lose wild type Kras2 during lung tumor progression.57 KRAS mutations more commonly occur in patients with a significant smoking history.58 Approximately, 70% of KRAS mutations are G→T transversions, with the substitution of glycine by either cysteine or valine. Similar G→T transversions are observed in the mutated p53 gene, representing similar DNA damage as a result of bulky DNA adducts caused by the polycyclic hydrocarbons and nitrosamines in tobacco smoke. This observation provides further evidence for a causative role for tobacco smoke in KRAS mutations. In a meta-analysis of 3,779 lung cancer patients, 18.4% of whom had KRAS mutations detected by polymerase chain reaction (PCR), mutant KRAS was associated with worse prognosis in adenocarcinoma but not in squamous histology.59 However, a prospective study failed to show a survival difference correlated with KRAS mutation in advanced NSCLC.60 Although the oncogenicity of aberrant KRAS mutation appears to be well established, all attempts to inhibit mutant KRAS have failed to demonstrate objective responses in clinical trials, including attempts to prevent binding to the inner cell membrane by inhibiting farnesyltransferase or geranylgeranyltransferase.61
Direct downstream effectors of RAS are the RAF1 and BRAF protooncogenes. Unlike RAS, mutations in the RAF1 gene have not been detected in human lung cancers. Its role is complex, as growth arrest of SCLC by activated RAF1 suggests that it has a TSG-like function, and one copy of RAF1 is frequently lost in lung cancer.62 Although BRAF, another member of the RAF family, is commonly mutated in malignant melanomas and colon cancers, mutations in this gene are found in fewer than 5% of lung cancers.
Activation of nuclear protooncogene products, such as those encoded by the myc family genes (MYC, MYCN, and MYCL) is often the end point for many signaling cascades. MYC, when heterodimerized with MAX, functions as a transcription factor, and this functional complex is necessary for normal cell cycle progression and differentiation, as well as programmed cell death. MYC gene amplification or transcriptional dysregulation is often observed in SCLC and to a lesser degree in NSCLC. One member of the MYC family is amplified in 18% of SCLC tumors and 31% of cell lines, compared to 8% of NSCLC tumors and 20% of cell lines. Amplification appears more frequently in SCLC patients previously treated with chemotherapy, giving rise to the “variant” subtype of SCLC, and its presence correlates with adverse survival.60 There are no MYC-specific drugs in development.
EML4-ALK fusion protein is a recently identified activating oncogenic driver of lung adenocarcinomas occurring in less than 5% of all NSCLC. It is formed by fusion of the N-terminal portion of the protein encoded by the echinoderm microtubule-associated protein-like 4 (EML4) gene with the intracellular signaling portion of the receptor tyrosine kinase encoded by the anaplastic lymphoma kinase (ALK) gene, resulting from a t(2:5) translocation.21 Multiple EML4-ALK variants have been identified in lung cancer.64–67 Similar to EGFR mutations, EML4-ALK fusions appear to occur almost exclusively in adenocarcinomas, specifically with acinar histology, in never or former light smokers.65,68–70 EML4-ALK fusions do not occur in tumors with mutations in EGFR or KRAS, and patients appear to be of younger age compared to patients with EGFR mutations.70 A variety of methods are currently being used to assess for the presence of the EML4-ALK fusion, including immunohistochemistry, FISH, and reverse transcription PCR (RT-PCR), however, each method has its own attributes and flaws and none has been adopted to date as a standard for testing. Several novel selective inhibitors of ALK kinase are in clinical development. A trial of crizotinib (PF-02341066), a dual MET/ALK inhibitor, in heavily pretreated patients with tumors that harbor the ALK fusion protein has resulted in a 57% response rate and 72% 6-month progression-free survival (PFS) with median PFS not reached at the time of report.71 Phase 2 and 3 trials with this agent are currently under way.
TUMOR SUPPRESSOR GENES AND GROWTH SUPPRESSION
A number of TSGs have been identified that inhibit lung tumorigenesis or suppress key phenotypes in developed lung carcinomas. Germline mutations in some TSGs such as LKB1, p53, and RB1 give rise to inherited tumor syndromes; however, somatic loss of TSGs within sporadic cancers is more commonly seen. Such genes can be lost through multiple mechanisms, such as inactivating mutations, chromosomal loss, methylation, or overexpression of other proteins that inhibit the suppressive gene’s expression or activity. Many classical tumor suppressors, such as the three identified above, have been identified by decades of work examining genes involved in human-inherited cancer syndromes and elucidating their role in sporadic cancers. Many additional suppressor gene candidates have been identified as a result of profiling large numbers of tumors for loss of heterozygosity (LOH) using high-resolution comparative genomic hybridization systematically to look for recurrent regions of chromosomal loss. Results are expected in the next few years from whole genome sequencing of hundreds of tumors to identify novel mutations.
p53 Pathway
The p53 gene, located at chromosome 17p13, is crucial for maintaining genomic integrity in the face of cellular stress from DNA damage through gamma and ultraviolet irradiation, carcinogens, and chemotherapy. It is the most frequently mutated TSG in human malignancies, and mutations affect approximately 90% of SCLCs and 50% of NSCLCs. In NSCLCs, p53 alterations occur more frequently in squamous cell (51%) and large cell (54%) carcinoma than adenocarcinomas (39%).72 p53 mutations have been linked to poorer prognosis retrospectively; however, they were not shown to correlate with survival in a prospective randomized clinical trial.60,73 Most p53 mutations are G→T transversions, which correlate with cigarette smoking. A major cigarette smoke carcinogen, benzo(a)pyrene, selectively forms adducts at p53 mutational hot spots.2 The Li-Fraumeni syndrome of inherited germline p53 mutation may also lead to increased susceptibility to lung cancer in adults; this risk is magnified by tobacco smoking, as carriers who smoked had a 3.16-fold higher risk for lung cancer than nonsmokers.74 The majority of missense mutations occur in the DNA-binding domain of the protein, and five of the six most prevalently mutated sites are arginine residues that are involved with electrostatic interactions with DNA strands.75 Missense mutations prolong the half-life of the p53 protein, leading to increased protein levels detectable with immunohistochemistry. Also, because p53 exerts its cellular actions as a tetramer, mutant forms of the protein appear to exert dominant negative effects on wild type p53 and have also been shown to inhibit the function of p63 and p73 family members.76
In addition to mutational or deletional loss of p53, other regulatory components of the p53 pathway are altered in lung cancer, including the ataxia-telangiectasia (ATM) gene, the p53 binding protein MDM2, and the p14ARF tumor suppressor. ATM and the related protein ATR are tumor suppressive serine/threonine kinases that activate cell cycle checkpoints in response to DNA damage and ultimately activate and stabilize p53.77 Although ATR and the downstream checkpoint kinases (CHEK) are mutated in less than 1% of lung adenocarcinomas, ATM has been found to have deleterious mutations in 7% of lung adenocarcinomas; these mutations were largely mutually exclusive with p53 mutations, likely indicating that mutations in both genes would have redundant effects, especially since it has been shown that gain-of-function mutations in p53 can inactivate ATM.78,79 Conversely, the MDM2 oncogene product negatively regulates p53 by binding its transcriptional activation domain, inducing its nuclear export, and by polyubiquitinating p53, marking it for proteasomal degradation.80 Abnormal overexpression of MDM2 is found in NSCLC, where it is amplified in a significant number of tumors. MDM2 activity is inhibited by other tumor suppressor genes in the p53 pathway, including ATM and also the p14ARF tumor suppressor gene, which is often lost in lung cancer.81
When p53 is functional, it is activated by phosphorylation in response to cellular stress (e.g., DNA damage). Once activated, p53 strongly induces the expression of other tumor suppressor genes that control cell cycle checkpoints (e.g., p21WAF1/CIP1), apoptosis (BAX), DNA repair (GADD45), and angiogenesis (thrombospondin).82 p53 activation has also been found to alter microRNA expression and maturation,83 and some effects of p53 are mediated by the induced expression of the microRNA miR-34.84 The high frequency of p53 loss across the entire spectrum of human tumors is a strong testament to its importance in inhibiting tumor development and growth. Restoring p53 activity in tumors is effective in halting their growth and could represent an effective therapy, although development of this strategy is challenging.85 Several gene therapy clinical trials have been reported in which lung cancers are treated by intratumoral injection (endobronchially or by computed tomography–guided needle injection), introducing a wild type p53 gene using retroviral or adenoviral vectors. Future therapeutic gains may come from combining gene and conventional therapies, but this approach may be limited to locoregional disease control.86 Other strategies have been developed to restore p53 activity without attempting to reintroduce the entire gene into tumor cells. These include the use of small molecule inhibitors of the MDM2–p53 interface that prevent the degradation of wild type p53 and the use of peptides and small molecules that are intended to restore wild type conformation to mutated p53.85,87 Immunologic targeting of cancer with vaccines is a different strategy that is potentially nontoxic and specific. Such a strategy takes advantage of novel protein sequences that result from p53 mutations, which can potentially be recognized as foreign epitopes by cytotoxic T cells mounting an immune response against a tumor. Vaccination of patients with advanced cancer with a custom vaccine corresponding to their tumor’s mutation in p53 or RAS has demonstrated the generation of mutant oncogene specific immune responses associated with prolonged survival.88 A trial of p53 vaccination in SCLC produced measurable increases in tumor-specific immune response that was associated with improved outcome and response to therapy in some patients.89,90
CYCLINS AND CELL CYCLE REGULATORY PATHWAYS
p16INK4A is a cyclin-dependent kinase (CDK) inhibitor important for the integrity of the G1 checkpoint. Loss of p16INK4A frees CDKs from inhibition, permitting constitutive phosphorylation of retinoblastoma (RB) protein and inactivation of its growth suppressive function. Approximately 40% of primary NSCLCs lose p16INK4A, located on chromosome 9p21, making it the most common component of the p16INK4A–cyclin D1–CDK4–RB pathway to be inactivated in NSCLC. Other CDK inhibitors are also lost at a lower prevalence in NSCLC, and the RB gene is mutated or lost in a significant minority of cases.79 In contrast, a strikingly different pattern of pathway dysregulation is observed in SCLC, in which abnormalities in p16 are rarely observed but RB itself is nearly always abnormal. Although p16INK4A point mutations in NSCLCs were observed in only 14% of tumors, homozygous deletions or aberrant promoter methylation are common mechanisms for p16INK4A inactivation.22 Indeed, aberrant p16INK4A methylation is a frequent, early preneoplastic event in the pathogenesis of squamous cell carcinomas.91 Furthermore, p16INK4A and p14ARF are alternative splice forms of RNA transcripts from the same DNA locus. p14ARF is also a tumor suppressor gene and functions to stabilize p53. Thus, alteration at the p16INK4A locus may not only abrogate p16INK4A function but also disrupt p53 pathway through p14ARF.92
In the absence of inhibitory regulation by the CDK inhibitors, cyclins and their catalytic partners, the CDKs, phosphorylate the retinoblastoma protein, a growth-suppressive nuclear phosphoprotein located on chromosomal region 13q14. When in its active, unphosphorylated form, RB binds and inactivates proteins such as transcription factor E2F-1 preventing G1/S transition. Mutations of one RB allele together with loss of the other wild type RB allele are frequently observed in SCLC. The RB protein is absent or structurally abnormal in more than 90% of SCLCs and 15% to 30% of NSCLCs. Lung-targeted, conditional deletion of Rb and p53 in mice leads to development of SCLC that recapitulated that observed in humans.93 Although the p16 and RB tumor suppressive components of this pathway are frequently lost in lung cancer, the growth promoting cyclin and CDK components of the pathway are often overexpressed and cyclin D1, cyclin E1, and CDK4 have each been shown to be amplified in a subset of lung cancers94 and are overexpressed by immunohistochemical evaluation.
LKB1, AMPK, mTOR Pathway
LKB1 is a serine/threonine kinase that serves as a “master regulator” of several key intracellular pathways through phosphorylation of downstream regulatory kinases. Its tumor suppressive role became apparent when it was discovered that inherited mutations in LKB1 gave rise to a rare autosomal dominant polyposis/cancer susceptibility disease, Peutz-Jeghers syndrome. This disease is characterized by the development of many hamartomas polyps throughout the intestinal tract, abnormalities in mucocutaneous pigmentation, and a 20-fold increase in lifetime risk for cancer, including gastrointestinal, breast, and pancreatic neoplasia.95,96 Subsequent to this discovery it was found that LKB1 is somatically mutated and deleted in a range of other carcinomas, most prevalently in non–small cell lung carcinoma, where approximately 20% to 30% of adenocarcinomas exhibit LKB1.97–99 Squamous cell and large cell lung carcinomas also exhibit LKB1 loss, but at a lower frequency, and immunohistochemical analysis revealed absent LKB1 expression in two-thirds of SCLC.100 In a mouse model, tumors rapidly develop when conditional Lkb1 knockout was combined with conditional expression of oncogenic Kras, using inhaled adenovirus-expressing Cre recombinase. High penetrance and multiple tumors per animal were noted. More than half of the incident tumors are associated with metastases. Additionally, whereas most other mouse models of lung tumorigenesis (e.g., oncogenic Kras with conditional p53 deletion) cause only lung adenocarcinomas, more than half of the tumors that result from Kras with conditional Lkb1 deletion showed squamous or mixed histology, and large cell histology was also observed.98
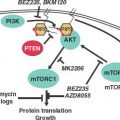
LKB1 regulates a key metabolic checkpoint through its phosphorylation of the AMP activated protein kinase AMPK. AMPK is sensitive to conditions of hypoxia and nutrient deprivation; under these conditions, AMPK is phosphorylated by LKB1, resulting in suppression of tumor growth and metabolic activity by direct phosphorylation of metabolic enzymes and by activation of the tuberous sclerosis complex tumor suppressors, which block activation of the mammalian target of rapamycin (mTOR) pathway.101 In tumors that have lost LKB1, this growth suppressive checkpoint is defunct. Resultant tumors show elevated activity of the mTOR pathway and may be selectively dependent on this pathway for growth, and pharmacologic activation of AMPK may also be a therapeutic target in these cancers.102,103 Metformin, an oral hypoglycemic drug commonly used for diabetes, activates AMPK and has been found to inhibit proliferation and colony formation in vitro.104 Retrospective analyses demonstrate reduced incidence of cancer among diabetics treated with metformin.105–107 In addition, metformin treatment for diabetes is associated with higher rates of complete response among neoadjuvantly treated breast cancer patients.108 Although metformin appears to require functional LKB1 in order to effect AMPK activation, other compounds have been identified that circumvent this requirement. Thus, direct pharmacologic reactivation of the downstream tumor suppressive functions of AMPK may be a viable therapeutic strategy for LKB1 deficient tumors.
In addition to its role in regulating the AMPK metabolic checkpoint, LKB1 has many other distinct roles dependent on other downstream effector kinases, such as salt-inducible kinase, NUAK, and microtubule-affinity regulating kinase. These actions play a role in regulating a variety of cellular phenotypes important to cancer, such as cellular motility and transcriptional regulation, and LKB1 has been shown to exert profound effects in maintaining cellular polarity.109,110 However, the relative importance of these various phenotypes in the biology of LKB1-deficient lung cancers is poorly understood.
Other Putative Tumor Suppressors
Several other genes that are less well characterized than the tumor suppressor genes detailed above have been identified as targets of recurrent mutational inactivation, chromosomal loss, and epigenetic repression in lung cancer. These candidate suppressor genes are often identified as regions of copy number loss or LOH that occur in multiple tumors in large genome-wide studies of chromosomal architecture in lung cancer. Further experimentation is required to elucidate which molecular pathways and cellular phenotypes are affected and to define the functional importance of these candidates. Common regions of genomic loss surround the chromosomal regions of classical tumor suppressors CDKN2A, CDKN2B, LKB1, and RB1 (in SCLC). Other areas of recurrent loss in NSCLC occur at 9p23, 3p14.2, 3p21.3 16q23.1, 2q21.2, 4q35, 5q12.1, and 13q12.11.94 Many of these regions are also altered in SCLC, although there are fewer data available for this tumor type. Areas of deletion may encompass many genes in any individual tumors. Determining the functional roles of each individual gene can be challenging. However, peak regions can be identified that are most frequently included in the deleted region across multiple tumors, and key genes are thought to be most likely to be included in these regions. Furthermore, integrating multiple types of data can reveal genes that are somatically inactivated by methylation or mutation, in addition to chromosomal loss; these are also likely to be the key suppressive genes within a deleted region. Of genes included in the regions listed above, missense mutations have been identified in PTPRD (9p23), LRP1B (2q21.2), BLU (3p21.3), and WWOX (16q23.1).111,112 Experimental reexpression of candidate genes has been shown to inhibit proliferation in tumor cell lines for many of these putative tumor suppressors, including PTPRD, LRP1B, WWOX (16q23.1), FHIT (3p14.2), SMARCA4 (19p13.2), PTEN (10q23), and RASSF1, FUS1, BLU, and SEMA3B (3p21.3).113,114 However, for most of these candidates the biological implications of gene loss in a tumor is uncertain. More research devoted to these targets is required to further elucidate the roles they may play in lung cancer biology with the goal of identifying driver pathways that may become activated in the absence of particular TSGs and may thus be effective targets for therapeutic intervention.
OTHER BIOLOGIC ABNORMALITIES IN LUNG CANCER
Cellular Immortality Resulting from Increased Telomerase Activity
Cellular senescence is mainly regulated by telomerase, a ribonucleoprotein enzyme responsible for maintaining telomere length by de novo synthesis of telomeres and elongation of existing telomeres. The human telomerase reverse transcriptase (hTERT) catalytic subunit is the major determinant of telomerase activity in vitro and in vivo. During normal cell division, telomere shortening leads to cell senescence and thus governs normal cell mortality. Telomerase reverse transcriptase maintains telomere ends via the synthesis of TTAGG nucleotide repeats. Telomerase activation is considered mandatory for tumor cells to escape senescence and contributes to immortalization and cancer pathogenesis. For example, immortalization of primary human airway epithelial cells can be achieved by the successive introduction of the simian virus SV40 early region and hTERT.115 Malignant transformation is seen when these immortalized cells are transfected by an activated RAS oncogene. Approximately 100% of SCLCs and 80% to 85% of NSCLCs have been demonstrated to express high levels of telomerase activity. Furthermore, hTERT gene amplification occurs in 57% of NSCLCs, suggesting that this pathway is commonly targeted in lung cancer.116 The prognostic significance of hTERT expression or activity remains controversial, although a recent study demonstrated that the copy number of serum hTERT mRNA was independently correlated with tumor size, tumor number, presence of metastasis, likelihood of recurrence, and smoking.117 Furthermore, elevated telomerase activity and hTERT levels have been associated with worse disease-free and overall survival in patients with stage I NSCLC.118 In preneoplastic lesions, telomerase activity or expression of its RNA component, or both, are observed in situ in lesions with the expression proportional to the severity of histology grade, supporting a temporal role for telomerase activation during lung preneoplasia.119 Thus, telomerase activity or expression can be used as a potential biomarker to detect premalignant as well as tumor cells. For these reasons, there is much interest in developing antitelomerase drugs as new therapeutics. GV1001 and HR2882 are telomerase peptide vaccines that are being evaluated in clinical trials in patients with NSCLC.120
Deregulation of Apoptosis
Loss of normal apoptosis commonly occurs in many cancer types and is associated with expansion of viable cells and the development of resistance to chemotherapy and radiation therapy. Many members of both the mitochondrial (intrinsic) and the death receptor (extrinsic) apoptotic signaling pathways are found to be abnormal in lung cancer. A member of the intrinsic pathway, the antiapoptotic gene BCL2 originally described in follicular lymphomas, is abnormally overexpressed in SCLC (75% to 95%) and some NSCLCs (25% of squamous cell carcinoma and approximately 10% of adenocarcinoma).121–123 BCL2 expression was associated with good prognosis in NSCLC.124 Cytotoxicity of many chemotherapeutic agents is induced through the BCL2 apoptotic pathway; overexpression of BCL2 is associated with increased resistance to these agents.125,126 Given the role of BCL2 in suppressing apoptosis and in reducing the efficacy of chemotherapy and radiotherapy, considerable effort is being made to develop BCL2-targeted therapeutics in combination with chemotherapy. In early phase 1 studies in patients with SCLC, BCL2 antisense was found to be well tolerated when combined with chemotherapy.127 However, randomized phase 2 trials found no difference in outcome compared with that of chemotherapy alone.128 Despite these discouraging results, a new class of oral BCL2 antagonists is current being developed.
In the extrinsic pathway, death receptors are members of the tumor necrosis factor (TNF) receptor gene superfamily that consists of more than 20 proteins with a broad range of biological functions, including regulation of cell death and survival, differentiation, or immune regulation. The best-characterized death receptor, Fas (CD95), and its ligand (FasL) have also been implicated in lung cancer. In general, lung cancers express FasL but not the receptor. However, as T cells express Fas, one model that may help explain the resistance of lung cancer cells to immune surveillance involves the clonal deletion of immune T cells that would otherwise be directed against lung cancer antigens by this Fas–FasL interaction. Both caspase-8 and caspase-10 expression appears to be decreased in lung cancer. Homozygous deletion or methylation of CASP8 gene has been observed in SCLC cell lines, with 79% demonstrating loss of expression.129 Polymorphisms in the promoter region of caspase-9 have been shown to contribute to risk of lung cancer development.130 Inhibitors of apoptosis (IAPs) impede cell death through caspase function, especially caspase-3 and -7. IAPs also inhibit apoptosis via modulation of the transcription of nuclear factor kB. One of the best-known members of this class of protein is survivin. Its expression is high in tumor but nearly nonexistent in adult normal tissue. Suppression of survivin has been shown to sensitize lung cancer cells to radiation, suggesting that it can be a potential target for intervention.131
Invasion, Metastasis, and Angiogenesis
Investigation of the molecular mechanisms of invasion and metastasis has yielded a variety of candidate genes, including cell adhesion molecules such as the cadherins, integrins, and CD44. The E-cadherin–catenin complex is critical for intercellular adhesiveness and maintenance of normal and malignant tissue architecture. Epigenetically reduced expression of this complex in malignant disease is associated with tumor invasion, metastasis, and unfavorable prognosis in lung cancer. Another family of adhesion molecules are the integrins. Integrin α3 has been shown to be important for normal lung development and diminished expression correlated with a poor prognosis of patients with lung adenocarcinoma. Specific isoforms of CD44 may also be associated with lung cancer metastasis. Matrix metalloproteinases (MMPs) are zinc-dependent proteases that belong to a family of endopeptidases, which degrade the extracellular matrix and basement membrane, necessary first steps in angiogenesis. Increased expression of MMPs has been strongly implicated in tumor growth, invasion, and metastasis. MMP2 and MMP9 have been associated with poorer prognosis. However, despite its established role in invasion and metastasis, many randomized phase 3 trials of MMP inhibitors have failed to demonstrate a survival benefit in patients with advanced lung cancer.132 This is perhaps related to the lack of specificity of these inhibitors and recent findings that some MMPs actually inhibit tumor growth.133 Many of the genes that confer invasive and metastatic phenotypes are coordinately regulated by transcription factors such as ZEB and SNAIL, which can be activated by several stimuli, especially the actions of transforming growth factor-beta.134 It has recently been shown that a key set of microRNAs of the miR-200 family plays a crucial role in regulating this phenotype.135 The stimuli that regulate tumor cell invasiveness are often generated or influenced by surrounding stromal or inflammatory cells, and as such, the invasive phenotype seems to be quite plastic, complex, and highly dependent on the context of the tumor microenvironment.136
Angiogenesis, the formation of new blood capillaries, is necessary for a tumor mass to grow beyond a few millimeters in size. The angiogenic switch results from perturbation in the balance between inducers and inhibitors, both of which are produced by tumor and host cells. Vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), and angiogenic cytokines, such as interleukin-8, have all been implicated in lung cancer.137 Furthermore, high microvessel density (MVD) and VEGF overexpression are predictive of poor outcome. Thus, tumor angiogenesis has become a major new therapeutic target for lung cancer.138 Clinical trials in lung cancer with agents targeting angiogenesis have shown great promise and have demonstrated that the addition of bevacizumab, the humanized monoclonal antibody to VEGF, to chemotherapy prolongs progression-free survival in phase 3 clinical trials, although improvements in overall survival were not always observed.139–141
CANCER STEM CELL HYPOTHESIS
The cancer stem cell hypothesis proposes that a self-renewing undifferentiated stem cell population that comprises a small fraction of the total tumor burden gives rise to more numerous and more differentiated progeny that populate the tumor.142 Among the characteristics reported to distinguish stem-like cell from cells constituting the bulk of tumors include the potential for supporting the continued growth of the local tumor mass, for seeding metastases throughout the body, and resistance to cytotoxic therapies that allow the residual viable stem cells to repopulate the tumor after treatment. Because of their resistance to treatment and potential for seeding distant metastatic disease, the study of cancer stem cell and development of strategies effectively to eradicate all residual stem cells is of critical importance in cancer treatment.
Cancer stem-like cells can be isolated from a variety of tumor types using antibodies to unique cell surface proteins. They are capable of forming xenograft tumors at a high frequency after injection into immunocompromised mice. Empirically selected surface markers have been used to isolate putative stem cells from human breast cancer, glioblastoma multiforme, colon cancer, and other carcinomas, and these cells have demonstrably greater potential for xenograft formation than do unselected tumor cells. The resulting tumors recapitulate the histological appearance of the primary tumor as well as the heterogeneous expression of various surface and intracellular molecular markers.142
Putative lung progenitor cells have been described as cells that reside at the bronchoalveolar duct junction that express both Clara cell and pneumocyte markers or as lung resident cells of hematopoietic origin that express CD133.143,144 These cells have not been conclusively shown to be lung adult stem cell populations but are intriguing and have been shown to be involved in repair of lung tissue after injury and may be involved in cancer development. In lung tumors, CD133145–148 and other commonly used markers of stem-like tumor cells—Hoechst dye efflux and aldehyde dehydrogenase activity149—have been shown to identify subsets of tumor cells that display characteristics consistent with the cancer stem cell hypothesis. CD133 has been shown to segregate with template DNA in lung cells undergoing asymmetric cell division.150 However, conflicting reports suggest that CD133 may not define a specific subset of cells, as interconversion between CD133+ and CD133− populations is observed, and CD133 expression may be associated with specific stages of cell cycle progression151; furthermore, some studies show no association between CD133 expression and propensity to initiate tumors.152 The cancer stem cell hypothesis has other important gray areas as well; it is not yet certain whether a consistent developmental hierarchy would exist for every individual tumor or only in certain cases; whether or not lineage differentiation in a tumor can be a reversible; and which basic properties should be required to define a stem-like phenotype in a given population of cells. A highly increased propensity for xenograft formation is one of the most convincing features that can be demonstrated experimentally for proposed cancer stem-like cells. However, even the reliability of this evidence is called into question by the observation that a much higher rate of tumor formation is observed when mouse tumor lines are propagated in isogenic immunocompetent mice, which raises the possibility that the xenograft initiation phenotype may be related to the ability to adapt to the tissue environment of an immunocompromised mouse, rather than a general property of enhanced tumor formation.153
Nevertheless, the balance of evidence from diverse tumor types favors the hypothesis that stem-like cells are present within tumors and may play a key role in certain aspects of tumor biology. Furthermore, developmental pathways that are proposed to be important in governing cancer stem cell biology may be important oncogenic drivers of proliferation and invasion for unselected tumor cell populations in certain subsets of tumors, and are important avenues of research in their own right. For instance, the activity of particular genes—achaete-scute complex homologue 1148 and OCT4 transcription factors146—have been implicated in regulating this subset of lung tumor cells. Given the far-reaching implications of the cancer stem cell hypothesis, and especially the concept that targeting developmental pathways cancer stem cells or in particular subsets of cancer could represent an important therapeutic strategy, these complex and exciting areas warrant further study in lung cancer.
GENOMIC ANALYSIS OF LUNG CANCER
In recent years technological advances in high-throughput sequencing approaches have enabled the comprehensive analyses of gene expression, copy number alterations, mutations, and other genetic perturbations across a large number of tumors. A number of studies have employed cDNA microarray chips to profile transcriptional expression in large sets of tumors. The largest such study of 443 lung adenocarcinomas from four institutions determined that clusters of coexpressed genes were consistently associated with patient outcome.154 Prognostic profiles have been derived in several independent studies, although it is unclear how reproducible their associations are when applied to independent datasets. Associations with patient prognosis and response to treatment are potentially useful as an adjunct to classical staging approaches to help inform clinical decision making. For instance, when treating early stage tumors, adjuvant treatment may be more likely to benefit patients with a poor prognostic profile; whereas, a good prognostic profile could justify avoidance of adjuvant therapy. Although gene expression profiling of breast cancers has been validated and are widely used for making adjuvant treatment decisions, so far gene expression assays in lung cancer have not been validated prospectively as a useful guide for treatment planning.
Beyond prognostic associations, global approaches to the study of lung cancer have great potential as discovery tools that can increase the understanding of cancer biology and the molecular mechanisms underlying key cancer phenotypes. Arguably the most important outcome of such studies will be the identification of subsets of tumors whose biology can be affected by interventions targeted against key dysregulated genes or pathways. Hypotheses regarding such relationships can be derived from statistical associations between patterns of transcriptional regulation, copy number alterations, and mutations and tumor characteristics such as proliferative rate, tendencies for invasion and metastasis, response to therapy, and survival. Several large studies have already begun in search of clinically important genetic patterns. High-resolution analysis of copy number alterations has led to the identification of chromosomal regions that are frequently amplified or deleted in subsets of lung cancer encompassing regions containing key oncogenes and tumor suppressor genes such as EGFR, KRAS, myc, p53, and LKB1, as well as other candidate genes whose importance may become apparent after further study.79,94 Microarray data from large cohorts of patients may reveal coordinately regulated sets of genes potentially associated with underlying dysregulation of particular pathways or cellular phenotypes. Such gene sets can be subjected to computational analyses to discover the biological and clinical significance (e.g., elucidation of specific oncogenic pathways, regulation of a transcription factors) inter alia. Furthermore, a greater depth of understanding may arise from coordinated analysis of multiple types of data from the same samples, greatly adding to the ability to interpret the significance of these data. For example, gene expression profiling has been carried out on tumors that have additionally been characterized by comparative genomic hybridization and extensive mutational sequencing112,155 or by microRNA expression profiling.156 It then becomes apparent that common mutations such p53 and EGFR, as well as regions of amplification and deletion, are consistently associated with altered expression of particular sets of genes.
A special case regarding the value of confluence of multiple types of data is seen with the analysis of large panels of lung cancer cell lines. Complete microarray expression profiling of the available lung cancer cell lines is complemented by independent knowledge of chromosomal changes, sequencing of mutations in common and novel cancer-associated genes, and profiling of microRNA expression, characterization of promoter methylation patterns and protein expression, and other molecular data. These molecular data can then be integrated with phenotypic observations such as sensitivity to targeted or cytotoxic therapies,157–159 pathway activation,160 or response to various perturbations.161 High throughput approaches have also been applied to genetic perturbations using short interfering RNA screens and to drug treatment of cell lines, and these data can give additional detail regarding functional significance of particular genes.162,163 Discovery efforts in cell lines may then generate hypotheses that can be applied back to enhance the understanding of data from primary tumors. For instance, expression profiles altered by oncogenic HRAS have been shown to be similar to profiles observed in lung cancer patients with KRAS mutations.160 Profiles thought to be associated with activation of PI3K seem to be up-regulated in normal bronchial epithelia from smokers with cancer compared with smokers without cancer.164 However, caution must be taken in generating these signatures and interpreting published associations, since a particular set of genes may be nonspecifically associated with the phenotype of interest, or the association may only be observed in the context of the in vitro system used in the experiments.
Global analysis of cancer biology is now poised to make an important step forward as massively parallel sequencing technologies become less expensive and more widespread. These technologies enable the sequencing of entire cancer genomes, allowing comprehensive determination of somatic mutations, polymorphisms, alternative splicing events, and chromosomal fusions. When this has been applied to a sufficient number of lung cancer samples, recurrent genetic alterations should become evident that may represent pharmaceutical targets or may highlight deregulated pathways where therapeutic intervention could be effective. The Cancer Genome Atlas represents a large-scale implementation of this strategy, and when it is complete it will combine gene expression, copy number alterations, single nucleotide polymorphisms, methylation status, microRNA expression, and mutation sequencing into a single compendium of data from several hundred clinically annotated tumors from multiple sites, including squamous cell lung carcinoma.
MOLECULAR TOOLS IN THE LUNG CANCER CLINIC
The understanding of the molecular genetic changes in lung cancer pathogenesis is advancing rapidly. Many genetic abnormalities identified in lung cancer are common to other human cancers, while others appear more specific for lung cancer, perhaps because of characteristics of the cells of origin and the unique nature of carcinogen exposure. Where their biochemical function is known, the proteins rendered abnormal appear to fall into several growth regulatory pathways.160 Thus, understanding of the fundamental workings and diverse molecular drivers of lung cancer is becoming clearer. A substantial effort has been made to translate the current scientific knowledge of these abnormalities from the bench to the bedside in order to improve patient outcomes. These approaches fall into three general categories:
1. Development of early detection tools to identify primary and recurrent disease to enable effective early treatment. Because lung cancer eventually develops in only one of ten cigarette smokers, the identification of persons with a genetic susceptibility to lung cancer should allow targeting and intensification of smoking cessation, early detection, and chemoprevention efforts. To date screening trials applied to smokers at high risk for the development of lung cancer, including the use of spiral computed tomography scans, have not been documented to decrease mortality. The identification of genetic epidemiologic markers and acquired respiratory genetic alterations may help to identify the most at-risk individuals for screening and chemoprevention trials.
2. Development of new cancer-specific therapies based on knowledge of genetic abnormalities. These may include replacing or pharmacologically reactivating mutant tumor suppressor genes, development of new drugs targeting activated protooncogenes, interfering with autocrine or paracrine growth stimulatory loops, and inhibiting angiogenesis, metastasis, and antiapoptotic. Some new therapies may be highly effective as single agents in some patients. However, it is likely that for many patients combinations of two or more targeted or cytotoxic agents will be required to maximize clinical benefit; and determining the optimal combination of therapy for a given patient will be an additional challenge in the field.
3. Identification of prognostic and predictive biomarkers, such as the EGFR mutation and ALK-EML4 fusion, previously described, that predict the response to specific therapies and prognosticate outcomes. Such tools will play an increasingly important role in selecting optimal treatment strategies as the number of molecularly targeted therapies expands.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


