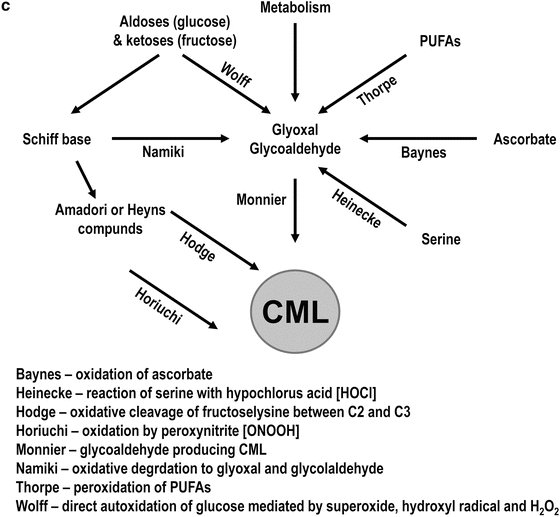
Fig. 8.1
Biochemistry of early and late glycation. (a) Early steps of the Maillard reaction. The reducing sugars in open chain form reacting an amino groups on proteins to form a reversible Schiff base. The Schiff base then form a cyclic glycosylamine or can rearrange to an enaminol and then to a ketoamine (Amadori compound). The Amadori compound is also stabilized by its cyclization to a furanose or pyranose ring. (b) The Amadori compound fructoselysine can undergo decomposition to form both 1- and 3-deoxyglucosone (1-DG and 3-DG). 3-DG is more reactive than glucose in the formation of AGEs. (c) Various pathways leading to the formation of AGEs. The Maillard pathway involves the reaction of a reducing sugar with an amine on a protein to form a ketoamine, which can break down to form AGEs. Alternatively, the autoxidation of glucose forms reactive compounds like arabinose and glyoxal that can further react with amino groups and form AGEs (Wolff pathway). The Schiff base intermediate can also form reactive carbonyl compounds under oxidizing conditions and can also react with an amine leading to AGE formation (Namiki pathway). Lastly, the ketoamine, under both oxidative and non-oxidative conditions, can fragment to form reactive deoxyosones that can form AGEs (Hodge pathway). Reproduced with modifications from: J.W. Baynes, “The role of AGEs in aging: causation or correlation”, Exp. Gerontol. (2001) 36(9), 1527–1537
In 1912 French food chemist Loius C. Maillard first described the formation of brown-colored substances from non-enzymatic reactions between reducing sugars and proteins [8], and such reactions are also relevant to the human body. A simplified view of the chemistry is that carbonyl groups and amino groups react to form Schiff bases and then Amadori compounds, (early glycation products), which are potentially reversible. Early glycation product formation may be followed by irreversible dehydration, condensation, and cross-linking reactions, resulting in a large, and likely incompletely known heterogeneous family of derivatives termed Advanced Glycation End-Products (AGEs). AGEs are also known as late glycation products, Maillard products, or glycoxidation products (as formation of many AGEs involves oxidative chemistry, see Fig. 8.2) [9]. Similar reactions can occur, by both enzymatic and non-enzymatic pathways, without glucose, providing the non-glucose materials contain an aldehyde group. Reactive metabolites such as the dicarbonyls (methylglyoxal (MG), glyoxal, and 3-deoxyglucosone (3DG)) from the glycolysis pathway, and from the metabolism of lipids and ketones can also interact with protein residues to form AGEs, including in lipoproteins [10]. Increased production of reactive dicarbonyls or their reduced detoxification by the glyoxalase system or by endogenous scavengers leads to increased carbonyl stress, which is a major driving force for AGE formation and accumulation [11]. AGE formation occurs in many extracellular and intracellular proteins, including lipoproteins, and AGEs are present in all people. AGE levels in long-lived tissues, such as skin, usually increase with chronologic age [12]. AGE formation is accelerated by hyperglycemia as in diabetes [13] and also by renal impairment, even in the non-diabetic milieu [14].
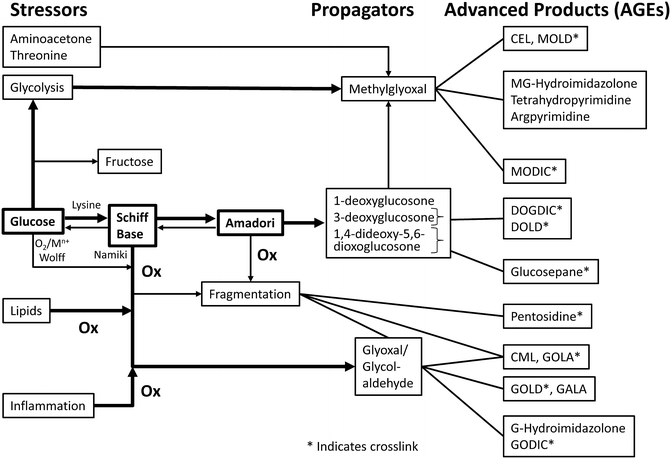
Fig. 8.2
Factors affecting AGEs formation and accumulation. Reproduced from: V.M. Monnier and X. Wu, “Enzymatic deglycation with amadoriase enzymes from Aspergillus sp. as a potential strategy against the complications of diabetes and aging” Biochem. Soc. Trans. (2003) 31, 1349–1353
AGEs are chemically heterogeneous groups of both fluorescent and nonfluorescent compounds with over 25 fully characterized AGE structures [15]. The (type and concentration) of glycation products formed depends on both the range and concentration of substrates available and the duration of their interaction. Nε-carboxymethyl-lysine (CML) is the simplest and best characterized AGE and the main epitope for many commercially available antibodies used for AGE detection and quantification. Many of these products such as CML (thought to be the most abundant AGE in vivo), pentosidine, and erythronic acid are formed oxidatively [16]. Non-oxidatively derived AGEs such as the imidazolones and pyrraline have also been identified and characterized [17, 18]. Pyrraline is formed by the reaction of 3-deoxyglucasone with lysine, and imidazolone-type AGEs are formed by the reaction of 3-deoxyglucasone with arginine. The value of each specific AGE, or group of AGEs, as a marker or mediator of diabetic microvascular and macrovascular damage is not fully elucidated.
AGEs can also be derived exogenously, such as from the diet and smoking [19–21]. Dietary AGEs, which are abundant in foods such as (all as per 100 g of product) fried pork bacon, roast chicken skin, sesame oil, parmesan cheese, sweet butter cream, pan fried beef or pizza [22]. AGEs in food are partially absorbed from the gastrointestinal tract, and approximately two-thirds are thought to remain in contact with tissues for several days, whereas the rest is rapidly excreted by the kidneys [23]. AGE restriction in mice, without energy or nutrient change, alleviates inflammation, prevents vascular complications, and extends their normal life span [24]. Human studies have showed that a low-AGE diet reduces inflammatory markers (C-reactive protein (CRP), Tumor Necrosis Factor alpha (TNFα)) and vascular cell adhesion molecule (VCAM-1) levels [25]. In Type 2 diabetes high-AGE meals have been shown to acutely impair vascular reactivity as measured by flow mediated dilation (FMD) [26]. HDL does suppress TNF-α induced VCAM-1 suppression in vitro, but it is not known how much of the low AGE diet benefit, in animals or in humans, relates to effects on AGE modified lipoproteins.
Differences Between Glycation and Glycosylation
The term “glycation” refers to non-enzymatic reactions between amino acid residues of proteins and reducing sugars. Glycosylation is a different set of usually enzymatic chemical reactions. Glycosylation is a major post-translational modification of both intracellular and extracellular proteins. Most intracellular proteins in humans contain sugars and are also known as glycoconjugates. Depending on the nature of the covalent attachment glycosylated proteins can be divided into glycoproteins (in which the major component is a protein) and proteoglycans (in which typically >95 % mass is a carbohydrate). Glycoproteins are an integral part of plasma membranes, and serve important functions such as hormones, receptors and mediators in intercellular interactions. Proteoglycans are major components of the extra cellular matrix (ECM) [27]. These ECM proteins can also become modified by (early and late) glycation, which is discussed elsewhere in this book.
Glycation of Apolipoproteins in Lipoproteins
Within lipoproteins apolipoproteins are major sites of glycation. Theoretically any amino compound with at least one hydrogen atom on its nitrogen can participate in the Maillard reaction. Chemically, within a protein moiety only amino acids with one or more nucleophilic residues (lysine (Lys), arginine (Arg), cysteine (Cys), methionine (Met), and histidine (His)) are likely to become glycated. Although the amino acid cysteine is the strongest nucleophile, Lys residues are particularly abundant in apolipoproteins [28] and thus are the preferred site of glycation. For example, ApoA-I, found in HDL, contains 243 amino acids residues, including three Met, 21 Lys, five His, and 16 Arg residues, but no Cys residues. ApoB-100, found in VLDL, LDL, and Lp(a), contains 4,563 amino acids residues: 79 Met, 356 Lys, 114 His, 150 Arg, and only 25 Cys (0.5 %). The extent of lipoprotein glycation will depend on (1) the time of lipoprotein exposure to the glycating agent, which may in turn be influenced by the location of the lipoprotein being glycated (e.g., intra- or extravascular); (2) the concentration of glycating agent; (3) the potency of the glycating agent; and (4) the efficacy of any deglycating or anti-glycating factors. The nature of the glycating agent determines the type of glycation products formed. For example protein glycation with glucose leads to the formation of the late glycation produce CML, whereas protein glycation with methylglyoxal results in formation of CEL [29]. In humans the major circulating glycating agent is glucose in an open chain form [27]. Circulating levels of glucose in non-diabetic subjects averaging at 5 mmol/l whilst that of methylglyoxal is 147 nmol/l [30], in addition several glycating agents may act on amino acids (in both the extracellular and intracellular milieu).
Extent of Lipoprotein Glycation
The extent of lipoprotein glycation usually correlates with other measures of glycemia such as HbA1c and fructosamine [4, 31, 32]. Any inconsistencies may relate to differences in half-lives of the glycated protein moieties, methodologies for the quantification of lipoprotein glycation (discussed below) and the range of glycemia related values in the study group. The half-life of lipoproteins is days, whilst HbA1c from within red blood cells, reflects glycemia over the previous 2–3 months, hence it is probable that the extent of lipoprotein glycation is more strongly correlated with shorter term measures of glycemia over days, such as mean glucose levels (perhaps measured by Continuous (Interstitial Fluid) Glucose Monitoring or blood glucose monitoring), or 1,5 anhydroglucitol levels [33], but such a comparative studies are not yet available.
The Measurement of Lipoprotein Glycation
The quantification of glycated lipoproteins is currently a research laboratory tool. Various techniques have been used, and have been predominantly applied to LDL and HDL. The most specific measure is the direct quantification of fructoselysine (an early glycation product) in lipoproteins by High Pressure Liquid Chromato-graphy (HPLC) [34], which requires the physical separation of lipoproteins by ultracentrifugation. We have utilized this technique to study lipoproteins from diabetes patients [35, 36].
Glycated proteins, such as albumin, and glycated lipoproteins bind to boronate, so boronate affinity chromatography has been used in both a preparative manner [37] and in a rapid relatively simple HPLC and gel permeation column based assay, developed by Tanaka et al. [38] which has been used to quantify glycated LDL and HDL from low volumes (5 μl) of serum.
Antibodies to glycated apoB have also been developed and used in in-house ELISA assays [39] and in a commercially available indirect competitive ELISA (Glyacor, Exocell, Philadel-phia, PA). A monoclonal antibody (ES12) is directed against a specific epitope in apoB in glycated LDL and does not cross-react with other human plasma proteins including non-glycated LDL. The assay range is 3–40 μg/ml (corresponding to 0.3–4 mg/dl) in serum. Other antibodies have also been used to quantify glycated HDL and glycated Lp(a) [40].
Unlike purely glycated unoxidized lipoproteins AGE modified lipoproteins have increased electrophoretic mobility [41], a technique usually used for the characterization of physically separated isolated lipoproteins. AGEs can also be quantified by Gas Chromatography/Mass Spectroscopy (GC/MS) [42] in separated lipoproteins. An AGE-LDL antibody based capture assay has also been developed [43] and used to quantify AGE-LDL in Type 1 diabetes.
A less specific biochemical tool to measure the extent of lipoprotein glycation is the TNBS (trinitrobenzene sulfonic acid) assay [44]. The TNBS assay measures the amount of free Lys in a protein. As mentioned earlier Lys is the most abundant amino group in human lipoproteins and is a strong nucleophile (Lys is the only one amino acid with two amino groups: alpha and epsilon). Unfortunately, due to the secondary and tertiary structure of proteins not all Lys residues (regardless of whether free or modified) are always available for reaction and therefore detection by the TNBS assay.
The development and validation of low cost high throughput assays of lipoprotein glycation would expedite this area of clinical research.
General Consequences of Lipoprotein Glycation
The potential consequences of increased lipoprotein glycation are summarized in Table 8.1. These include effects on lipoprotein metabolism (such as on their half-life in the circulation) and on cell interactions and responses, including effects related to important processes (e.g., inflammation, thrombosis, vasoreactivity) relevant to the vascular complications of diabetes. Lipoproteins modified by glycation and by oxidation and extravasated are more likely to bind to vascular matrix, such as proteoglycans, than unmodified lipoproteins [45]. Tsmikas et al. demonstrated that the concentration of oxidized LDL in the arterial wall is 70-fold that in the circulation [46], but we are not aware of similar studies related to glycated lipoproteins. Matrix binding of lipoproteins is discussed in more detail in another chapter.
Table 8.1
Adverse effects of lipoprotein glycation
Effects on circulating half-life of lipoproteins |
Foam cell formation |
Increased matrix binding |
Pro-oxidant and reduction of antioxidant effects |
Pro-inflammatory or reduced anti-inflammatory effects |
Pro-thrombotic effects |
Pro-apoptotic effects |
Effects on lipoprotein related enzyme activities |
Altered lipoprotein receptor interactions |
Cell signaling effects |
Effects on gene expression |
Promotion of antibody and immune complex formation |
Altered reactivity in assays |
It is important to recognize that even normoglycemic people have some lipoproteins that undergo glycation, and that more extensively modified (late glycation or AGE modified) lipoproteins, may not remain in the circulation very long. Indeed AGE-modified lipoproteins are likely to exist predominantly outside the potent antioxidant milieu of blood in the extravascular spaces (of arteries, the retina, and renal beds), being rapidly removed from the circulation by pathways such as scavenger receptors in liver and in white blood cells. Antioxidants in blood include albumin, urate, bilirubin, and vitamin C [47], all of which are water soluble. Some fat soluble vitamins, which can be carried within the lipoproteins (e.g., Vitamin E) are also antioxidants [48]. The low concentrations of modified lipoproteins in the circulation (relative to unmodified lipoproteins) may reflect both that formed within blood and that has effluxed from the extravascular bed.
Another challenge in this area of research is that in vitro modified lipoproteins studied in the laboratory setting may be differentially or more extensively modified than that occurring in vivo. Often the glucose or reactive intermediate (e.g., methylglyoxal) concentrations and incubation times used in the laboratory are well beyond that present in people. Later in this chapter we will point out some studies in which both in vivo and in vitro glycated lipoproteins were studied, with divergent responses.
In the literature related to in vitro modified lipoproteins the term glycation is often used loosely, not specifying if it is early or late glycation. The effects of early glycation and late glycation often differ. For example, in in vitro studies of modified LDL on cultured retinal or renal cells by Lyons et al. both LDL modified by early glycation (glycated LDL) and LDL modified by late glycation (Heavily oxidized glycated LDL (HOG-LDL)) have been studied. HOG-LDL effects were generally significantly greater than that of less extensively modified glycated LDL [49–52]. Ideally researchers should present data related to the preparation and characterization of the modified lipoproteins they have studied. The in vitro modification of lipoproteins by early glycation alone requires the presence of metal chelating antioxidants, such as EDTA and DTPA in adequate concentrations and reduced exposure to oxygen such as may be achieved by incubation under nitrogen or argon and dialysis against nitrogen purged buffers [7]. In general, if there is increased electrophoretic mobility of lipoproteins on agarose gels, or increased AGEs or lack of recognition of modified LDL by the classical LDL receptor, then the glycation is more advanced (late glycation).
Whilst in vivo studies, including longitudinal human or animal studies, can also be informative as to the effects of lipoprotein glycation, we must evaluate their findings whilst also recognizing that improved glycemic control may use lifestyle changes and drugs which may have direct effects on lipoprotein related gene or protein expression or other pleiotropic effects, and that more than just glycemia (and lipoprotein glycation) may change. Some of the particularly relevant sites of change, such as within the vascular wall, in the retina or within glomeruli or renal tubules, may not be accessible for sampling, particularly in living humans.
Human Studies of Glycated Lipoproteins
Glycated lipoproteins, particularly those modified by early glycation, are present in the circulation of both non-diabetic and diabetic people at relatively high concentrations [53, 54] Durrington et al. have demonstrated that circulating levels of glycated apoB (which may reflect glycated apolipoprotein B within LDL, VLDL, Lp(a), and chylomicrons) are increased in primary conditions in which LDL is raised, such as heterozygous familial hypercholesterolemia. As with hyperglycemia itself, the hallmark of diabetes mellitus, enhanced lipoprotein glycation occurs from diabetes onset, and likely reflects both an increase in the number of glycated amino acids per lipoprotein particle and also a greater proportion of lipoprotein particles with glycated residues. Based on our studies of in vivo glycated LDL as assessed by boronate affinity chromatography, in non-diabetic subjects approximately 5 % of LDL particles are sufficiently glycated to bind to these columns (and have increased fructoselysine levels) whereas in people with diabetes (depending on their level of glycemic control) up to 25 % of LDL may bind [35, 37]. Even within an individual the extent of glycation of lipoproteins will likely vary, in the same way that not all LDL, HDL or VLDL particles are the same size [55, 56]. At any given time the circulating lipoproteins will include some that are newly secreted, hence are likely to be less glycated, and lipoproteins that are several days older, hence more likely to be more glycated. Ambient glucose levels and lipoprotein size, apolipoprotein content and chemical composition are also likely to affect the extent of lipoprotein glycation. For example, Durrington et al. demonstrated that small LDL is more likely to undergo in vitro glycation than larger LDL [54].
Glycation of Specific Major Lipoprotein Classes
VLDL Glycation
Whilst hypertriglyceridemia is common in people with Type 2 diabetes and in those with Type 1 diabetes and poor glycemic control, obesity or renal damage there are few studies of VLDL glycation.
Levels of Glycated VLDL
Using a simple and non-specific agarose gel electrophoresis assay for glycated lipoproteins in sera from diabetic and non-diabetic subjects levels of glycated VLDL were estimated to be fourfold higher in diabetes subjects and higher in diabetic patients with vs. without clinically evident atherosclerosis [57].
Effects on Lipoprotein Metabolism
Hypertriglyceridemia may relate to both increased hepatic VLDL production and delayed VLDL clearance. In keeping, in in vivo VLDL kinetic studies in rodents the clearance of triglyceride and apoB of in vitro glycated VLDL was slower than that from normal VLDL. and in in vitro studies the glycated VLDL was a poorer substrate for lipoprotein lipase [58].
There are several studies comparing VLDL from diabetic subjects and non-diabetic subjects which demonstrate that VLDL from people with Type 1 and Type 2 diabetes has a different lipid and apolipoprotein content from that of non-diabetic subjects, and within the same Type 1 or Type 2 diabetic patient can differ when their glycemic control is poor vs. improved, and is associated with increased rates of cholesteryl ester synthesis by human monocyte derived macrophages [59–61] and endothelial cells [59–62]. Levels of or the extent of VLDL glycation were not quantified in these studies of modified VLDL.
LDL Glycation
Studies of LDL glycation are more numerous than those of other lipoprotein fractions, likely because LDL is highly atherogenic, especially when modified, and is usually the most abundant lipoprotein in blood and in atherosclerotic plaque.
Levels of Glycation
Relative to non-diabetic people the levels of glycated LDL are increased (by approximately 50 % to several fold) in Type 1 and Type 2 diabetes subjects, and usually correlate with other measures of glycemia or with LDL-C levels, two required substrates for LDL glycation [39, 55, 63–65]. In Type 2 diabetes patients levels of AGE-LDL were also elevated (about three-fold) relative to non-diabetic subjects, and were lower in diabetic patients taking metformin than in those not on metformin [65]. Levels of circulating glycated LDL have been found to be higher in Type 2 diabetic patients fed a high AGE diet than in low AGE diet fed diabetic patients and non-diabetic subjects [66].
LDL Size and Glycation
Small dense LDL is more atherogenic than larger more buoyant LDL particles [4]. There are divergent results from studies relating LDL size and LDL glycation. Glycated LDL (in the absence of LDL antibodies) has a longer residence time in the circulation [67], thus may be smaller due to further lipolysis and lipid exchange. By evaluating in vivo modified and in vitro glycated LDL particles some studies have suggested that small dense LDL is more susceptible to glycation [54, 68]. Isolated LDL modified in vitro with methylgloxal to form AGE-LDL was also significantly smaller than the native LDL [69]. However, in Type 1 diabetic patients, using NMR spectroscopy we found no significant difference in the size of in vivo glycated and relatively non-glycated LDL separated by boronate affinity chromatography [35].
Susceptibility to Oxidation
Oxidized LDL is more pathogenic than unmodified (native) LDL. Results of studies of the effects of LDL glycation on LDL’s susceptibility to oxidation are divergent, perhaps related to differences between in vivo and in vitro modification, the type, concentration and exposure time to the pro-oxidant, and the assays used to quantify oxidation. Tsai et al. demonstrated increased susceptibility of LDL from Type 1 diabetic patients with poor glycemic control to in vitro (copper-induced) oxidation [70]. This was not so in our study of complication-free Type 1 diabetic subjects with relatively good glycemic control [71]. We also determined the in vitro susceptibility to copper induced oxidation of relatively glycated LDL (G-LDL) and relatively non-glycated LDL (G-LDL) prepared by boronate affinity chromatography from 13 subjects with Type 1 diabetes. Lipid soluble antioxidant levels did not differ between the two subfractions, in keeping with a lack of increased oxidative stress to G-LDL in plasma. The lag time to oxidation of the G-LDL was significantly less than that of the non-glycated LDL subfraction. There were no significant differences in the rate of or extent of lipid oxidation during the reaction, nor did the lag time, rate or extent of protein oxidation of the two LDL subfractions differ [35]. In cross-sectional analyses of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) cohort we did not observe any statistically significant relationship between LDL susceptibility to lipid or protein oxidation and HbA1c and severity of nephropathy or retinopathy [72].
Glycated LDL and Immune Complex Formation
Antibodies to, and immune complexes with, modified lipoproteins such as glycated and AGE-modified LDL are implicated in human vascular damage. Modified lipoproteins themselves are pro-inflammatory, but when in immune complexes they are even more pathogenic. Such immune complexes can increase foam cell formation and have pro-inflammatory effects, features of microvascular complications as well as atherosclerosis [73]. In Type 1 diabetes subjects of the DCCT/EDIC cohort levels of AGE-LDL in circulating immune complexes are associated with and predict progression of carotid intima-media thickness [74], and also predict progression of diabetic retinopathy [75].
Matrix Binding
Lipoprotein matrix interactions, also discussed in another book chapter, may promote atherosclerosis and may also accelerate diabetic nephropathy by binding to glomerular matrix and affecting renal cell signaling [76]. Matrix binding and retention of LDL and of glycated and/or oxidized LDL is thought to increase LDL’s likelihood of further modification by glycation, oxidation and AGE formation.
In vitro generated AGE-LDL has been found to be smaller and to bind more avidly to proteoglycans [69]. Using an in vitro model system of binding to arterial wall proteoglycans Edwards et al. demonstrated that improved glycemic control in Type 2 diabetes patients reduced LDL proteoglycan binding, even in the absence of significant improvements in lipid levels. LDL glycation (fructosamine) was the only LDL compositional variable that correlated (r = 0.95) with the proteoglycan binding [77].
Effects on Receptor Interactions and Cell Signaling
Lipoprotein glycation can change LDL’s cell-based receptors and responsive cell signaling pathways in cells relevant to the vascular complications of diabetes. In general LDL modified by early glycation can still interact with the classical LDL receptor on cells, as does unmodified (native) LDL, but with increasing degrees of glycation major pathways of cellular uptake are via scavenger receptors, the Receptor for AGEs (RAGE), and by endocytosis [49, 78–80].
Glycated-LDL was isolated from diabetic patients and from non-diabetic subjects. In cultured human fibroblasts, which express only the classical LDL receptor the rates of receptor-mediated accumulation of relatively non-glycated LDL from both subject groups were greater than those of glycated-LDL. In contrast, when incubated with human monocyte-derived macrophages, the rates of receptor-mediated accumulation of glycated-LDL from both groups were significantly greater than those of non-glycated-LDL [37].
We exposed cultured rat mesangial cells to native LDL or to LDL modified (in vitro) by early glycation or by extensive oxidation and glycation (AGE-LDL). Glycated LDL was taken up via the classical LDL receptor, induced a transient intracellular calcium spike and marked extracellular signal–regulated protein kinase (ERK) activation. AGE-LDL, recognized by the scavenger receptor, induced a sustained rise in intracellular calcium and less marked ERK activation [49]. In cultured human vascular smooth muscle cells relative to native LDL AGE-LDL significantly increased protein and/or gene expression of receptors for modified LDL and AGE proteins (LRP1, CD36 and RAGE), which was associated with adverse cellular responses related to oxidative stress and cell proliferation [79].
Adverse Cellular Effects
Early and late glycation of LDL has been demonstrated to have many adverse cellular effects which may promote macro- and microvascular damage in diabetes. Most studies involve cultured monocytes, or arterial, retinal and glomerular cells exposed to in vivo or in vitro glycated LDL. Adverse cellular responses include foam cell formation, cell proliferation or death (commonly by apoptosis), matrix overproduction (of particular relevance to glomerulosclerosis), pro-inflammatory effects and (discussed in subsequent sections in this chapter) impaired vasorelaxation and pro-thrombotic effects.
Macrophages are implicated in atherosclerosis and also in diabetic microvascular damage. Lopes-Virella et al. demonstrated than human monocyte derived macrophage had increased cholesteryl ester accumulation when exposed to in vivo modified LDL from diabetic subjects, or to in vitro glycated LDL [31, 36]. Several groups demonstrated increased cholesterol uptake and cholesteryl ester accumulation by macrophages in response to glycated LDL, with greater effects of more extensively modified LDL, such as AGE-LDL generated by glycoaldehyde [80–82].
In cultured human vascular endothelial cells (HUVEC) in vivo and in vitro glycated LDL can induce apoptosis [83] and in vitro generated AGE-LDL can increase expression of monocyte chemoattractant protein (MCP) [84], which may also promote atheroma. AGE-LDL induced MCP-1 expression in cultured human endothelial cells has been demonstrated to be ameliorated by the PPARα agonist lipid drug fenofibrate, and by the anti-platelet agent dilazep, both of which suppressed the AGE-LDL induction of NFκB [85].
With regard to cultured microvascular cells, Lyons et al. demonstrated reduced cell viability of retinal capillary cells after exposure to in vitro glycated vs. native LDL [86] and reduction in this cytotoxicity by LDL glycation in the presence of the AGE inhibitor aminoguanidine [87].
Whilst we found that glycated LDL did not reduce mesangial cell viability, it increased mesangial cell TGFβ mRNA expression and induced hemeoxygenase-1 (HO-1) expression, an intracellular marker of oxidative stress (personal communication A Jenkins). Others have demonstrated altered mesangial cell modified LDL binding and increased matrix (e.g., fibronectin and laminin) production by cultured mesangial cells exposed to glycated LDL than to native LDL [76, 88–91]. These changes may promote glomerulosclerosis, a major feature of diabetic nephropathy.
Effects on Modulators of Fibrinolysis
Exposure of cultured human vascular endothelial cells to in vitro glycated LDL increases PAI-1 production [92, 93]. This process is via activation of the PAI promotor [94] and involves the Golgi apparatus [95] and RAGE [96], and decreases generation of tissue plasminogen activator (tPA) [93]. In contrast, using in vivo modified LDL from Type 1 diabetic subjects separated by boronate affinity chromatography into glycated and relatively non-glycated LDL subfractions PAI-1 and tPA production by human aortic endothelial cells exposed did not differ significantly [35]. The different responses may relate to different extents of LDL glycation and cell types.
Effects on Platelet Reactivity
Platelet hyperactivation is a common feature of diabetes and may promote thromboses in both large and small vessels. LDL that was AGE modified in vitro and LDL from Type 2 diabetic patients with poor glycemic control stimulated platelet p38MAPK phosphorylation and thromboxane B2 production [97].
Another group demonstrated that relative to native LDL in vitro glycated LDL increased platelet TBARS levels (a measure of oxidative damage), NO production, intracellular calcium levels, and ADP-induced aggregation [98].
Effects on Vasoreactivity
Glycated LDL can also impair vascular reactivity. Whilst early glycation of LDL (without oxidation) had no effect on aortic ring acetylcholine-induced endothelium dependent relaxation AGE-modified LDL attenuated their vasorelaxation to an even greater extent than Ox-LDL [99]. In keeping with these results, AGE-LDL impaired acetylcholine-induced endothelium-dependent vasorelaxation of isolated mouse aortas, which was prevented by pharmacological inhibition of calpain. Exposure of bovine aortic endothelial cells to this same type of AGE-LDL reduced eNOS protein levels in a dose and time-dependent manner, without altering eNOS mRNA levels, increased intracellular calcium and reactive oxygen species production [100].
In cultured porcine aortic endothelial cells exposed to in vivo glycated LDL and relatively non-glycated LDL from diabetic and non-diabetic subjects (separated by boronate affinity chromatography) the glycated LDL increased superoxide release by five-fold relative to the non-glycated LDL [101].
Both in vivo modified LDL from diabetic patients and in vitro glycated LDL caused vasoconstriction of arterioles in skeletal muscle of living mice [102], in keeping with similar adverse effects on vascular tone in the microvasculature.
HDL Glycation
Glycation of HDL in diabetes may ameliorate the efficacy of some of HDL’s vasoprotective functions, which include reverse cholesterol transport, antioxidant, anti-inflammatory, anti-thrombotic, and vasodilatory effects. As with other lipoprotein subclasses, there is an admixture of studies using in vivo and in vitro modified HDL, and some studies of in vitro modified HDL use glycating agent concentrations or incubation times which may not occur in vivo.
Levels of Glycated HDL
Relative to that in non-diabetic subjects the level of glycation of HDL is increased about four-fold in people with Type 1 or Type 2 diabetes and correlates with other measures of glycemic control. Whilst all HDL associated apolipoproteins are glycated, about 80 % of HDL glycation is located on ApoA1. In in vitro studies for any given glucose concentration the extent of apoA1 glycation was significantly greater in the presence of phospholipids [103].
Antioxidant effects of HDL can be assessed by measuring the susceptibility to oxidation and also the efficacy of HDL in breaking down preformed lipid peroxides. Oxidation is implicated in the formation of late glycation (AGE) products, which also occur in HDL. The literature is divergent as to the effects of HDL glycation of its susceptibility to oxidation, which may relate to different oxidation techniques and measures of oxidation.
Using 50 mM d-glucose, aluminum, and iron, one group demonstrated increased oxidative damage in HDL [104], whilst another group found that glycated HDL was less, not more susceptible to in vitro oxidation by copper based on a xylenol orange assay, with no difference in levels of induced conjugated dienes or thiobarbituric acid reactive substances (TBARS) [105].
In Type 2 diabetic patients with diabetic nephropathy serum AGE levels were increased and isolated (in vivo modified) HDL was less effective than that from nondiabetic subjects in protecting against ex vivo LDL oxidation (induced by DCFH), however the extent of HDL glycation was not reported [106].
Using in vivo and in vitro modified HDL and oxidized red blood cell (RBC) membranes we found that the efficacy of HDL to remove preformed lipid peroxides (LPO) from RBC membranes was significantly impaired with HDL from complication-free Type 1 diabetes patients relative to healthy subjects. We did not quantify HDL glycation, but relative to unmodified HDL in vitro glycated HDL from non-diabetic subjects did not have impaired LPO removal efficacy, whilst AGE-modified HDL did, suggesting that late but not early glycation may be deleterious [107]. In a similar model system HDL from Type 2 diabetes patients with in vivo glycated paraxonase-1 (PON-1) was less able to break down preformed LPO, with in vitro AGE modification having greater function effects on this HDL function than in vitro HDL glycation [108].
Effects on Modulators of Fibrinolysis
In people with diabetes circulating levels of PAI-1 are often increased, and in cultured vascular endothelial cells Shen et al. demonstrated that glycated HDL increased HUVEC PAI-1 production, whilst unmodified HDL had no effect. Neither native nor glycated HDL altered endothelial cell tPA production [93, 94]; however, in HUVEC cell culture the effects of HDL from non-diabetic and diabetic patients on tPA or PAI-1 production were similar. If HDL glycation has such an effect in vivo this could promote thrombosis.
Effects on Vasoreactivity
HDL can have vasodilatory effects. In a rabbit aortic ring model HDL from Type 1 diabetic patients could not attenuate the inhibitory effects of Oxidized LDL on endothelial dependent vasodilatation as well as HDL from non-diabetic subjects. However, this effect was not correlated with HDL-fructosamine levels (reflecting HDL glycation) or other systemic measures of glycemia [109].
Reverse Cholesterol Transport
The transport of cholesterol from cells to HDL is one of the more well-known functions of HDL. Results of studies related to the effects of HDL glycation are divergent, which again may reflect the extent of HDL glycation and the model systems used.
In general reverse cholesterol transport is thought to be impaired in people with Type 2 diabetes and in mouse models, but some investigators have reported greater cholesterol efflux with Type 2 diabetic HDL than non-diabtetic HDL, but no measures of HDL glycation were reported [110]. In a model of cholesterol efflux from mouse peritoneal macrophages HDL from Type 1 diabetes had impaired cholesterol efflux, but this did not correlate with measures of HDL glycation, nor was the function of in vitro glycated HDL impaired [111]. In another study of in vitro glycated HDL its ability to promote cholesterol efflux was not significantly altered [105].
In an in vivo model of macrophage-to-feces RCT HDL mediated cholesterol efflux was reduced (about 20 %) in Type 1 diabetic rodents vs. non-diabtetic rodents, with unchanged cholesterol efflux to diabetic HDL but lower SR-BI mediated uptake from Type 1 diabetic HDL. Both in vitro and in vivo experiments supported effects due to HDL glycation [112].
Anti-inflammatory Effects
Another role of HDL is inhibition of vascular endothelial cell adhesion molecule expression (CAMs), such as VCAM-1 and ICAM [113, 114]. CAMs promote the attachment, rolling, and ingress of monocytes into the vascular wall, and levels of circulating forms, such as soluble (s) VCAM-1, sICAM, and sE-selectin, are increased in Type 1 and Type 2 diabetes [115], and circulating CAM levels have been correlated with circulating HDL-C levels, but correlations with glycated HDL levels have not been reported. CAM expression is also implicated in diabetic nephropathy [116] and diabetic retinopathy [117] and serum levels can be acutely lowered by intensive insulin treatment [118], but levels of glycated HDL were not reported. In our rabbit studies of collared carotid arteries the suppression of vascular CAMs was attenuated by methylglyoxal glycated ApoA1 and by ApoA1 from diabetic patients relative to unmodified ApoA1. The collars caused intima/media neutrophil infiltration and increased endothelial expression of VCAM-1) and ICAM-1. Unmodified ApoA1 infusions decreased neutrophil infiltration and CAM expression substantially, whilst in vitro glycated ApoA1 was less effective at suppressing neutrophil infiltration and did not significantly lower CAM expression. The in vivo glycated ApoA-I from diabetic patients did not inhibit neutrophil infiltration or CAM expression. These reduced anti-inflammatory properties of glycated ApoA1 were related to reduced inhibition of NFκB and reactive oxygen species (ROS) formation [119].
In keeping, another group demonstrated that in vitro glycated and AGE modified HDL, with increased levels of both fructoselysine and carboxymethylysine, had reduced PON activity and did not suppress oxidized LDL induced monocyte adhesion to human aortic endothelial cells, as did unmodified ApoA1 [120]. In contrast in vitro glycation of HDL did not impair its ability to inhibit monocyte adhesion to cultured aortic endothelial cells [120].
Perhaps also related to CAM expression glycated HDL increased breast cell adhesion to HUVEC and to extracellular matrix, implicating HDL glycation in cancer metastasis [121].
In another model of inflammation, of high glucose induced redox signaling in human monocyte-derived macrophages, apoA1 inhibited glucose-induced oxidative stress (ROS generation, NADPH expression, Nox2, SOD 1 and superoxide production) whilst in vitro glycated apoA1 and that from Type 2 diabetic subjects was less effective [122]. In THP1 cells, human monocyte derived macrophages and mouse RAW2647 cells native HDL suppresses lipopolysaccharide (LPS) induced cytokine (TNFα and interleukin-1β(IL-1β) release, whilst in vitro (28-fold) and in vivo (4-fold) glycated HDL were significantly less effective [123].
Lipoprotein (a) Glycation
The pro-atherogenic and pro-thrombotic lipoprotein lipoprotein (a) (Lp(a)), which is discussed in another book chapter, also undergoes non-enzymatic glycation in diabetes, and this may enhance its adverse vascular effects.
Levels of Glycated Lp(a)
In a small cross-sectional study using boronate affinity chromatography and immunonephelometry serum levels of glycated Lp(a) were found to be increased (more than double) in Type 2 diabetes patients relative to non-diabetic subjects, and higher in those with vs. without diabetes complications, but the extent of apoB glycation within Lp(a) was relatively higher [124]. In keeping, Doucet et al. demonstrated (using boronate affinity chromatography and ELISA) that levels of glycated Lp(a) were about 50 % higher in diabetic than non-diabetic patients, with apo(a) being less glycated than the apoB within Lp(a). Glycated Lp(a) levels correlated positively with HbA1c levels. Their in vitro glycation studies demonstrated that Lp(a) was less susceptible to non-enzymatic glycation by glucose than LDL [40].
Susceptibility to Oxidation
As usually found with LDL, glycation of Lp(a) increases its susceptibility to in vitro copper induced oxidation [125].
Effects on Modulators of Fibrinolysis
Relative to native Lp(a), glycation (including late glycation) of Lp(a) increases the production of PAI-1 and PAI-1 mRNA expression in cultured HUVEC and human coronary artery endothelial cells and suppresses tPA synthesis and secretion (but not mRNA expression). These changes are attenuated by the AGE inhibitor aminoguanidine and by the lipid soluble antioxidant butylated hydroxytoluene (BHT) [126], implicating the importance of combined glycation and oxidation (AGE modification). These changes may impair fibrinolysis and promote vascular thrombosis and clinically evident vascular events.
Effects on Vascular Reactivity
In people with diabetes vascular reactivity is usually impaired, contributed to by reduced nitric oxide (NO) bioavailability (which is also discussed elsewhere in this book). In a model system of isolated rat aortic rings glycated Lp(a) without concomitant oxidation did not impair acetylcholine (Ach)-induced endothelium dependent vasodilation, whilst oxidized Lp(a) and AGE modified Lp(a) did, with AGE-Lp(a) having the most deleterious effects. The likely mechanism is by increased superoxide formation, which may inactivate NO [125].
Glycation of Lipoprotein Related Enzymes
Lipoprotein related enzymes, found on the lipoproteins themselves and on cells with which they interact, mediate exchange of constituents between lipoproteins, alter lipoprotein composition (e.g., by cholesterol esterification), and have antioxidant effects. Glycation may affect these enzymes directly by modification of their amino acid components, by altering their reactivity with their glycation modified lipoprotein substrates or receptors, or by a combination thereof. The role of altered activity of these enzymes due to glycation and their potential as a therapeutic target for amelioration of diabetes vascular complications has not been fully delineated.
We now review studies of the effects of glycation on some important lipoprotein related enzymes, including Platelet Activating Factor Acetylhydrolase (PAFAH), located mainly on LDL, and of paraoxonase, Lecithin-Cholesterol Acyl Transferase (LCAT), and Cholesteryl Ester Transfer Protein (CETP), which are predominantly located on HDL.
Platelet Activating Factor Acetylhydrolase (PAFAH)
The enzyme PAFAH, which is also known as lipoprotein-associated phospholipase A(2) hydrolyzes and inactivates the lipid mediator Platelet-Activating Factor (PAF) and/or oxidized phospholipids. PAF is a phospholipid that activates neutrophils, macrophages, platelets, and smooth muscle cells, and increases vascular cell adhesion molecule (CAM) expression and vascular permeability. Increased PAF and/or decreased PAFAH levels or activity have been associated with atherosclerosis and inflammation [127]. PAFAH, circulates on LDL and to a lesser extent on HDL, and can inhibit lipoprotein oxidation [127, 128], but there are few studies of the effects of lipoprotein glycation on PAFAH. Serum PAFAH activity levels have been found to be increased in people with Type 1 diabetes [129–131] and with Type 2 diabetes [132] relative to non-diabetic subjects, and to be increased in renal failure [133], perhaps as a compensatory protective response. PAFAH activity in diabetes correlated with LDL-C and HDL-C levels in both forms of diabetes [129–132, 134] and correlated inversely with HbA1c levels in Type 1 diabetes [129]. Whilst serum PAFAH activity in Type 1 diabetes correlates with LDL susceptibility to oxidation and with Oxidized LDL levels [130, 134] the relationships between lipoprotein glycation and PAFAH are not yet reported.
Paraoxonase (PON)
There are three PON genes and related proteins [135]. PON1 and PON3 proteins are located on HDL and have protective effects against LDL oxidation. PON2 is also implicated in vascular damage in diabetes [136], but is not known to be associated with lipoproteins. The glycoprotein PON1 is predominantly synthesized in the liver, is located in tissues, in particular the kidney [137, 138] and in serum is located exclusively on HDL [139], with a preference for certain apo J containing and smaller HDL subclasses [140, 141]. PON protects against exogenous organophosphate poisons and in vivo is thought to hydrolyze phospholipid oxidation products [137], homocysteine, thiolactone [142], “statins” [143] and to protect against modifications of lipoproteins and cell membranes. Acute-phase HDL is less protective against LDL oxidation: this type of HDL has greatly reduced PON1 activity [144]. PON1 activity is usually assessed in vitro by hydrolysis of the artificial substrates of paraoxon and phenylacetate [137] and lactones [145].
A major determinant of PON activity are PON genotypes, which have also been associated with cardiovascular disease in the general [137, 146] and diabetic [147, 148] populations, and with diabetic retinopathy and nephropathy [149–151]. PON genotype may also modulate glycemia in both non-diabetic [152] and diabetic subjects [153, 154], which in turn may affect glycation of all lipoprotein classes.
PON protein levels are usually normal in diabetes [155, 156], but there is reduced serum PON activity in people with Type 1 and Type 2 diabetes [150, 155, 156]. In some cross-sectional studies serum PON activity is lower in diabetic subjects with neuropathy [157], retinopathy [158] and nephropathy [159], but not in others [153]. PON activity in humans can be increased by statins and fibrates [135, 160].
Mackness et al. postulate that the low PON1 activity observed in diabetes is due to non-enzymatic glycation [153], which is in keeping with in vitro studies [156] or a circulating inhibitor of PON [155]. HbA1c and serum PON activity were not well correlated in our cross-sectional studies [150], but this may relate to differences in half-lives. Shorter term measures of glycemia (e.g., glucose records over a few days) are preferable because they correspond more closely to the (several days) half-life of PON. Longitudinal studies of improved glycemic control and PON activity and lipoprotein glycation are desirable.
Lecithin: Cholesterol Acyl Transferase (LCAT), a glycoprotein produced by the liver, is preferentially bound to circulating HDL, and is also found on VLDL and LDL [113]. LCAT which catalyzes esterification of free cholesterol to cholesteryl ester, and may also hydrolyze oxidized lipids, is the rate-limiting enzyme in reverse cholesterol transport [161]. LCAT activity is inhibited by HDL2, lipid peroxidation products [162–164], and activated by apoAI and ApoAIV, both of which may become glycated. LCAT activity is decreased in both Type 1 and Type 2 diabetes [165, 166] and in uremia [167]. Whilst some have found that LCAT activity and glycemia do not correlate in diabetes [168] Nakhjavani et al. found that LCAT activity and HbA1c were negatively correlated (rho = 0.951) in Type 2 diabetes subjects, and on multivariate analysis HbA1c was a strong independent predictor of LCAT activity. LCAT activity and Oxidized LDL levels in serum also correlated, but relationships between LCAT and glycated lipoproteins were not reported [169]. In longitudinal studies LCAT activity decreases with glycemia improved by insulin [170, 171], but not by diet or sulfonylureas [170].
In 1995 Fournier et al. reported both in vivo and in vitro modified LCAT and its reactivity to non-diabetic and diabetic (in vivo glycated) HDL [172]. The kinetics of isolated non-diabetic LCAT activity varied according to the extent of in vitro LCAT glycation. Moderate glycation (<30 % residues on the TNBS reactivity assay) increased Km and Vmax, whilst greater glycation reduced both Km and Vmax. At all levels of LCAT glycation the LCAT reactivity was lower in the presence of in vitro glycated HDL, related to the extent of lysine glycation in (the potent LCAT activator) apoA1. With in vivo modified HDL (from diabetic patients) as LCAT substrate Km values were not altered, but Vmax and LCAT reactivity were reduced by about 30 %. These differences between in vitro and in vivo glycated HDL may relate to physiochemical changes other than glycation. More recently in in vitro studies Nobecourt et al. demonstrated that methylgloxal-induced late glycation of apoA1 impaired its ability to activate LCAT, which was ameliorated by the late glycation inhibitors aminoguanidine and pyridoxamine, the AGE breaker alagebrium, and the insulin sensitizer metformin [114].
Cholesteryl Ester Transfer Protein (CETP)
CETP, a glycoprotein, stimulates transfer of cholesteryl ester, triglycerides and phospholipids between circulating lipoproteins, such that triglyceride-rich lipoproteins lose triglyceride and gain cholesteryl esters [113], and is a key enzyme in reverse cholesterol transport [173] Synthesized by hepatocytes, adipose tissue and arterial smooth muscle cells [174] CETP binds to VLDL, LDL and HDL. CETP gene polymorphisms influence HDL levels and vascular disease [175]. The effects of glycemia and lipoprotein glycation on CETP activity have been studied. CETP activity is increased in people with Type 1 [176] and Type 2 diabetes [177] relative to non-diabetic subjects. In diabetes patients subcutaneous insulin delivery activates, while intraperitoneal delivery reduces, CETP activity [178]. Glycemia may influence CETP activity via non-enzymatic glycation of the enzyme [179] and via conformational changes which affect enzyme binding and lipid exchange. Passarelli et al. showed that in vitro glycated and in vivo glycated lipoproteins are associated with increased cholesteryl ester transfer rates from HDL to VLDL and LDL. Whilst in vitro glycation of partially purified CETP markedly impaired its activity [179], greater lipid transfer rates were observed when in vivo glycated lipoproteins from diabetic subjects were used, which was attributed to glycation of HDL protein. Lemkadem et al. demonstrated that in vitro glycation of HDL3 (with glucose concentrations up to 200 mM) increased cholesteryl ester transfer, but kinetic studies showed a paradoxical increase in CETP activity associated with a decrease of CETP affinity. HDL lipid and protein composition was unchanged but its fluidity was decreased and its electronegativity increased, which may affect CETP reactivity [180].
Treatment of Lipoprotein Glycation in Diabetes
Approaches that may reduce lipoprotein glycation are listed in Table 8.2. The general approaches include reduction in “substrate stress” by lowering levels of glucose (and other glycating agents) and of lipids, the inhibition of early and late glycation reactions, the use of deglycating agents and the removal of existent AGEs. Another strategy would be to modulate adverse cellular and extracellular matrix responses to glycated lipoproteins.
Table 8.2
Potential approaches to reduce lipoprotein glycation
Lower glucose levels |
Lifestyle, e.g., diets such as low AGE diets |
Glucose control drugs, e.g., metformin, insulin, sulfonylureas, incretins |
Lower lipid levels |
Lifestyle |
Drugs such as statins, fibrates, ezetimibe, resins |
LDL apharesis |
Combined glucose and lipid lowering drugs, e.g., colestamide |
Inhibit glycation reactions |
Early glycation, e.g., saponins, some nutrients |
Late glycation, e.g., amadorins |
Removal of preformed AGEs |
AGE breakers? |
Deglycating drugs |
Increase activity of deglycating enzymes |
Lipid control. Dyslipidemia, even by the traditional narrower definition related to measures of lipid levels, is a common associate of diabetes, in particular in the setting of poor glycemic control. As discussed elsewhere in this book, improving the lipid profile is an important aspect of preventing the macro- and microvascular complications of diabetes. Improved glycemic control, weight control, a healthy diet, exercise and non-smoking status are important goals which will also improve the lipid profile, but often, particularly in developed countries, lipid drugs are required. The benefits of the major lipid drug classes (statins and fibrates) for cardiovascular, retinal and renal event reductions have been shown in prospective placebo controlled randomized clinical trials, predominantly in Type 2 diabetes [183–188]. These benefits likely relate to both direct lipid lowering effects and pleiotropic effects
Improved glycemic control, which also reduces diabetic vascular complications [161, 189–191] will improve the traditional lipid profile and also reduce post-translational lipoprotein glycation, reducing substrate stress. Unfortunately in clinical practice achieving normoglycemia is often challenging related to availability, affordability, and efficacy of current glucose control drugs and insulin pumps, and patient and clinician fears of hypoglycemia, which may also have adverse cardiovascular effects. Strategies that can reduce lipoprotein glycation, or the adverse cellular and enzymatic responses to lipoprotein glycation, even in the setting of hyperglycemia, are desirable. Apart from glucose lowering drugs and perhaps HMG CoA reductase inhibitors (statins) [192] there are currently no therapies in clinical practice known to reduce lipoprotein glycation.
Glucose control agents: As discussed above several prospective longitudinal studies have demonstrated that drugs, such as insulin and metformin which improve glucose control, are associated with reduction in levels of glycated lipoproteins [193–195]. Some, such as metformin, may also have pleiotropic effects such as antioxidant or anti-AGE effects [196]. This is most likely related to effects on lowering ambient glucose levels and related improvements in the lipid profile. There is a novel class of glucose lowering agents, Sodium Glucose Transporter 2 inhibitors, which induce glycosuria via inhibition of glucose reabsorption by the renal tubules, which are currently in human clinical trials [197, 198] and are now approved in some countries for clinical use. As yet there are no published studies related to their effects on lipoprotein glycation.
Lipid drugs that are commonly used in clinical practice, such as the statins and fibrates, effectively improve dyslipidemia in diabetes, reduce the risk of macro- and microvascular events [199, 200], and also have pleiotropic antioxidant effects [201, 202] but there are few publications of effects on lipoprotein glycation. Whilst recent studies suggest that statins may increase Type 2 diabetes risk [203] and in a small prospective Type 2 diabetes study atorvastatin worsened glycemia [204] there is no definitive evidence that statins, fibrates or ezetemibe substantially alter circulating glucose levels in people with diabetes, which could impact lipoprotein glycation. In a cross-sectional study Younis et al. demonstrated lower levels of plasma glycated apoB in statin-treated type 2 diabetes patients compared with those not on statins [205]. This may relate to changes in LDL levels rather than a direct effect on lipoprotein glycation. Longitudinal studies are merited. Lipid drugs such as the statins and fibrates may also affect lipoprotein related enzyme activities, as discussed earlier.
The anion exchange resin colestimide improves both glycemia and lipid levels in people with Type 2 diabetes, hence could be expected to reduce lipoprotein glycation, but as yet there are no related publications [204, 206]. Conversely, nicotinic acid, particularly the rapid release preparations, whilst improving the lipid profile (in particular lowering VLDL and increasing HDL levels), can slightly worsen glycemia [207], so may increase lipoprotein glycation, but as yet there are no relevant published data of glycated lipoprotein levels.
LDL Apharesis. LDL apharesis, originally used for the treatment of familial hyperlipidemia, has been used for the treatment of patients with peripheral vascular disease, the nephrotic syndrome due to steroid-resistant focal glomerulosclerosis (FGS) and diabetic nephropathy. LDL apharesis (also discussed in another chapter) effectively lowers LDL and Lp(a) levels in people with diabetes, and has been shown to lower circulating levels of malondialdehyde (MDA) modified (oxidized) LDL, but again, there are no studies of the effects of apharesis on levels of glycated LDL (and other glycated lipoproteins). Interestingly LDL apharesis has been shown to lower levels of an intracellular glycated protein (Hb) (HbA1c), so it may be expected to lower glycated lipoprotein levels. LDL apharesis also has many other favorable effects beyond lipid-lowering, including improving blood viscosity, platelet aggregation, anti-inflammatory effects, vasodilatory effects and increases in (pro-angiogenic) Vascular Endothelial Growth Factor (VEGF) levels [208–211].
Related posts:
 Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
 Insulin Resistance and Atherosclerosis
Niacin Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
Insulin Resistance and Atherosclerosis
Niacin Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
 Lipoproteins and Diabetic Nephropathy
Lipoproteins and Diabetic Nephropathy
 Fibrate Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
Fibrate Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
 Apoproteins and Cell Surface Receptors Regulating Lipoprotein Metabolism in the Setting of Type 2 Diabetes
Apoproteins and Cell Surface Receptors Regulating Lipoprotein Metabolism in the Setting of Type 2 Diabetes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


