Leukocyte Function and Contribution to Disease
Edith Van De Vijver
Taco W. Kuijpers
Inflammation is a tightly regulated process initiated after tissue injury or infection. The main function of inflammation is to eliminate the pathogenic insult and remove damaged tissue, with the aim of restoring tissue homeostasis. The concerted action of professional phagocytes—neutrophils, monocytes, and macrophages—is crucial to the effective elimination of intruders and cell debris.
Neutrophils are short-living cells that are recruited early in the inflammatory response. Upon binding of cytokines, which originate either from microorganisms or from surrounding cells, neutrophils transmigrate through the vascular wall toward the infected tissue. The opsonized pathogen is bound and phagocytosed by the neutrophil, whereupon it is killed by mediators such as toxic granular proteins or reactive oxygen species (ROS). These inflammatory mechanisms are potentially harmful to the host, with the risk of inducing sepsis. Therefore, neutrophil activation has to be tightly controlled by several negative regulation mechanisms.
Inflammation also shifts the hemostatic mechanisms in favor of thrombosis. Multiple mechanisms are at play, including upregulation of tissue factor leading to the initiation of clotting and amplification of the clotting process by augmenting exposure of cellular coagulant phospholipids. Moreover, a decrease in the activity of natural anticoagulant pathways takes place, which is particularly targeted toward downregulation of the activated protein C (APC) pathway through multiple mechanisms. The decreased function of the natural coagulation pathways appears to play a role in dampening inflammatory responses. The dual roles of the neutrophil in inflammatory responses and hemostasis are highlighted in this chapter.
DEVELOPMENT AND TURNOVER
About 60% of all nucleated cells in the bone marrow belong to the myeloid series. Neutrophils originate from myeloid progenitor cells, and they mature in the bone marrow in about 2 weeks. During maturation and until activation of neutrophils, it is important that the neutrophils are retained in the bone marrow. Interactions between chemokine receptor CXCR4 on neutrophils and its ligand CXCL12, which is constitutively expressed by bone marrow stromal cells, play a crucial role in this process. Disruption of this interaction by administering CXCR4 antagonist AMD3100 results in neutrophilia due to increased neutrophil release from the bone marrow.1
During myelopoiesis, which includes the development of granulocytic as well as monocytic lineages, the myeloidspecific growth factors, granulocyte colony-stimulating factor (G-CSF) and granulocyte-monocyte-CSF (GM-CSF), play an important role.2 G-CSF and GM-CSF exert a dual effect: a boost in cell survival as well as the induction of myeloid development through an increase in the number of colony-forming units. The effect of G-CSF is transmitted to the myeloid cell through the G-CSF receptor. Mice deficient in either G-CSF or its receptor have a reduced neutrophil count (˜20% of normal).3,4
Normally, <2% of all neutrophils in the body are circulating in the blood. When released from the bone marrow, neutrophils normally have a short life span (6 to 10 hours) in the bloodstream.5 Thereafter, they move to the extravascular tissues and within hours undergo spontaneous apoptosis. Indeed, neutrophils are predisposed to cell death by apoptosis. This process prevents the cytotoxic contents in neutrophil granules from being released into the surrounding tissues, and it facilitates the elimination of dead neutrophils by tissue macrophages.
The exact molecular mechanisms underlying neutrophil apoptosis are unknown, although members of the Bcl-2 protein family and caspases have been shown to be involved.6 At the level of mitochondria, these two groups of proteins are intimately connected: Bcl-2 homologues govern the activity of caspases by exerting their effect through regulation of mitochondrial function.7 Proapoptotic Bcl-2 proteins, such as Bax, redistribute from the cytosol to the mitochondria to disturb the mitochondrial membrane integrity by forming channels, which facilitates the subsequent release of cytochrome c and the activation of Apaf-1 and downstream caspases. The family of caspase proteases executes cleavage of specific targets, which then leads to cell disassembly and death. Among these proteases, caspase-3 stands out for the large number of substrates that it destroys, including nuclear proteins, cytoplasmic structures, and cytoskeletal elements.
In the clinic, survival of both the immature myeloid progenitor cells and mature neutrophils can be extended by delaying apoptosis through the action of a wide variety of agents, including G-CSF and GM-CSF. During infections, the release of neutrophils into the circulation is accelerated, and these cells are also among the first to leave the blood vessels by extravasation and to migrate toward the site of infection.
EXTRAVASATION AND MIGRATION
During inflammation, circulating neutrophils migrate to the site of infection following a gradient of chemotaxins in a process called chemotaxis. Chemotaxins may be derived from either the infected tissue or local complement activation, or directly from the pathogens themselves, and diffuse within the tissue into the local vasculature. These gradients of chemotaxins recruit the neutrophils in interplay with factors expressed on the
luminal side of blood vessel endothelial cells locally.8 Many of the chemotaxins involved in granulocyte movement are small proteins of about 60 to 100 amino acids, very homologous in structure. The major family of chemotaxins, known as the chemokine superfamily (see Chapter 46), consists of over 30 different chemotactic molecules, which are produced by host cells in response to inflammation, injury, hypoxia, or other forms of stress. Most chemokines can be classified into α (CXC) and β (CC) chemokines, distinguished by the presence or absence of a single amino acid between the first two of four conserved cysteines.9 The γ (single C) and δ (CX3C) classes of chemokines have only recently been coined, each with as yet one member. As exemplified by CXCL8—the first CXC member described— most CXC chemokines activate neutrophils, whereas the CC chemokines act toward various lymphocyte subsets, monocytes, eosinophils, or basophils. CXCL8 is also referred to as interleukin-8 (IL-8).
luminal side of blood vessel endothelial cells locally.8 Many of the chemotaxins involved in granulocyte movement are small proteins of about 60 to 100 amino acids, very homologous in structure. The major family of chemotaxins, known as the chemokine superfamily (see Chapter 46), consists of over 30 different chemotactic molecules, which are produced by host cells in response to inflammation, injury, hypoxia, or other forms of stress. Most chemokines can be classified into α (CXC) and β (CC) chemokines, distinguished by the presence or absence of a single amino acid between the first two of four conserved cysteines.9 The γ (single C) and δ (CX3C) classes of chemokines have only recently been coined, each with as yet one member. As exemplified by CXCL8—the first CXC member described— most CXC chemokines activate neutrophils, whereas the CC chemokines act toward various lymphocyte subsets, monocytes, eosinophils, or basophils. CXCL8 is also referred to as interleukin-8 (IL-8).
In addition to the chemokines, chemotactic peptides are released by infecting microorganisms, for example, formylmethionyl-leucyl-phenylalanine, and the host complement system, for example, the split product of activated C5 (C5a). Cell membrane-derived lipid mediators, such as platelet activating factor, are also strong chemoattractants. For each of these agents, specific receptors on the neutrophil plasma membrane exist. These receptors, as well as those for the chemokines, belong to the seven-span superfamily of G-protein-coupled receptors (GPCRs), which are integral membrane proteins with seven transmembrane domains. Ligand specificity is created by differences in the extracellular domains. The intracellular domains interact with trimeric guanidine triphosphate (GTP)-binding proteins, enabling a link with signal transduction pathways leading to a wide range of functional responses, the complexity of which is still largely unclear.
Cells following the chemotaxin gradient toward the site of infection have to leave the bloodstream in a process called extravasation (FIGURE 47.1). Extravasation is a multistep process involving adhesion molecules in which chemotaxins function as activating agents or (pro-) inflammatory mediators. The first step of extravasation consists of initial contact between endothelial cells and neutrophils marginated by the fluid flow of the blood. L-Selectin (CD62L) on leukocytes plays a role herein, contacting several cell adhesion molecules on endothelial cells. Within the local environment of an inflammatory tissue reaction, the endothelium begins to express the adhesion molecules P-selectin (CD62P) and later on E-selectin (CD62E) (see Chapter 46).10 The low-avidity interaction of these selectins with their ligands on the opposite cells forces the neutrophils to slow down and make a rolling movement along the vessel wall.
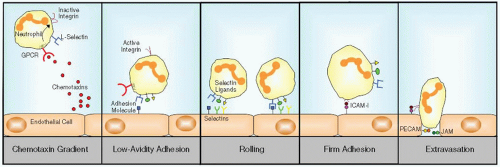 FIGURE 47.1 Extravasation of neutrophils. Neutrophils migrate in the direction of a gradient of chemotaxins, released at the site of infection. Upon initial contact with the endothelial cells through L-selectin, neutrophils slow down and interact with endothelial cell selectins while rolling along the vessel wall. Subsequently, integrins on the neutrophil are activated and bind to adhesion molecules on the endothelial cells, which results in stable adhesion. Binding to PECAM, JAMs, and other adhesion molecules guides neutrophils through the endothelial layer in the direction of the infection. |
In contrast to the low-avidity binding of neutrophils to selectins, the final step of firm adhesion and subsequent migration depends on stable interaction between integrins on the neutrophil and their ligands on the endothelial cells. For this process, the integrins need to be “activated,” that is, change their conformation into one with high binding avidity for their ligands, which is described in more detail in Chapter 46. Integrins consist of heterodimeric proteins of an α-chain covalently associated with a β-chain.11 One particular β-chain may associate with one of various α-chains. As a consequence, several integrin receptor subfamilies exist. The β2 integrin receptor subfamily is only expressed on leukocytes and comprises four different heterodimeric proteins, each of which contains a different α subunit: that is, αLβ2 (LFA-1; CD11a/CD18), αMβ2 (CR3; CD11b/CD18), αXβ2 (gp150,95; CD11c/CD18), and αDβ2 (CD11d/CD18). The β2 integrins bind to adhesion molecules on endothelial cells (intercellular adhesion molecule [ICAM]-1 and ICAM-2) and to several complement factors. The main β2 integrin on neutrophils is CR3.
In contrast to rolling and adhesion of neutrophils, the mechanisms that mediate migration through venular walls are less well understood. However, there is now good evidence that besides ICAM-I and -II junctional molecules such as PECAM-1 (CD31) and the junctional adhesion molecule (JAM) family (JAM-A, -B, and -C) play a major role.12,13
As mentioned above, slowly rolling neutrophils are able to recognize concentration differences in a gradient of chemotaxins and to direct their movement toward the source of these
agents. Although the details of this process remain unknown, the gradient mostly likely causes a difference in the number of chemotaxin receptors on either side of the cell, thereby inducing the cytoskeletal rearrangements needed for movement. Since adhesion molecules such as the β2 integrins are essential for the connections with the tissue cells or with the extracellular matrix proteins, these connections must be formed at the front of the moving neutrophils and broken at the rear end.8,14 Moreover, for continued sensing of the chemotaxin gradient, the chemotaxin receptors on the neutrophil must be freed from their ligand for repeated usage. This occurs through internalization of the ligand-receptor complex, intracellular disruption of the connection, and transport of the free receptor to the front of the cell, followed by reappearance of the free receptor on the leukocyte surface. Within the infected tissue, the chemotaxin gradient persists, and leukocyte migration is maintained.
agents. Although the details of this process remain unknown, the gradient mostly likely causes a difference in the number of chemotaxin receptors on either side of the cell, thereby inducing the cytoskeletal rearrangements needed for movement. Since adhesion molecules such as the β2 integrins are essential for the connections with the tissue cells or with the extracellular matrix proteins, these connections must be formed at the front of the moving neutrophils and broken at the rear end.8,14 Moreover, for continued sensing of the chemotaxin gradient, the chemotaxin receptors on the neutrophil must be freed from their ligand for repeated usage. This occurs through internalization of the ligand-receptor complex, intracellular disruption of the connection, and transport of the free receptor to the front of the cell, followed by reappearance of the free receptor on the leukocyte surface. Within the infected tissue, the chemotaxin gradient persists, and leukocyte migration is maintained.
INTRACELLULAR SIGNALING
This complex process of cytoskeletal remodeling and cellular movement is controlled by several small GTPases of the Rho family and their guanine-nucleotide exchange factors (GEFs). In neutrophils, it has been shown that, in particular, Rac-2 is crucial in the signaling leading to cytoskeletal remodeling and cellular motility. Deficiency in Rac-2 or its upstream regulator PI3Kγ results in failure to chemotax and to degranulate upon GPCR activation. P-Rex1, a GEF for Rac, is believed to be regulated by Gβγ and PtdIns(3,4,5)P3 signaling and thereby links GPCRs and PI3Kγ activity to Rac-dependent neutrophil responses.15
Several other GEFs, for example, DOCK2 and the Vav proteins, are also important for cell movement. Upon GPCR stimulation, neutrophils deficient for DOCK2 show impaired chemotaxis due to loss of polarization of the cell.16 Vav1/3 knock-out neutrophils, on the other hand, show defective β2 integrin-induced responses, which are specifically required for stable adhesion.17
Recent studies have suggested that GTPase Rap1 is also involved in neutrophil adhesion. We were the first to describe a syndrome of combined leukocyte adhesion deficiency and platelet dysfunction despite normal integrin expression, designated as LAD-1/variant syndrome.18,19 In a boy with a similar clinical syndrome, then named LAD-III, Rap1 activity was found to be absent.20 Therefore, the Rap1 regulation by CalDAG-GEF1 was suspected to be defective in these clinical pictures.21 However, recently we and others showed mutations in FERMT3, encoding a hematopoietic structural protein called kindlin-3, as the genetic cause for this syndrome. Kindlin-3 acts as a switch to activate the integrins similar to what was known for talin at the time.22,23 Whether and how exactly these two proteins interact is still unclear.24
IL17-PRODUCING CELLS AS A DRIVING FORCE BEHIND NEUTROPHILIA
Neutrophil release from the bone marrow into the blood stream has been extensively studied, and depends to a great extent on cytokines.25 An important group of cytokines in this respect is the IL family, which is mainly produced by activated lymphocytes and macrophages. ILs stimulate other immune cells to differentiate and proliferate, and can induce fever. The identification of ILs, as well as discoveries of their receptors, and their production have markedly changed the way in which we study immunologic processes. Through the study of the role of IL-23 in autoimmunity, an alternative T cell subset was discovered: the adaptive Th17 cell, whose expression is promoted by both chronic inflammation and tissue damage.26 The Th17 cell was identified as a cell that induces an inflammatory response by producing IL-17, IL-21, and IL-22, and is thereby clearly distinct from the well known Th1 and Th2 subsets. Furthermore, Th17 cells have been associated with autoimmune diseases.27
Interestingly, the IL-17-mediated immune pathway is induced within 4 to 8 hours following epithelial cell injury or activation of pattern recognition receptors (PRRs), which is too short to allow the development of Th17 cells. In vivo models of infection have shown that several innate cell subsets, among which unconventional γδ T cells, cooperate to promote this immediate IL-17-mediated response. Studies of extracellular pathogens that target the lungs showed that IL-23 was a main stimulator of innate IL-17 production.28 Innate IL-17— producing cells activate epithelial cells to release granulopoietic factors such as G-CSF, thereby recruiting large numbers of neutrophils crucial for effective and rapid control of bacterial and fungal pathogens. Furthermore, IL-17 synergizes with other inflammatory cytokines, such as IL-1, IL-6, and tumor necrosis factor (TNF)-α.
It is notable that IL-17 often works cooperatively with IL-22, during homeostasis as well as during pathogenic insults. In the airways, IL-17 and IL-22 have been shown to work together to promote the secretion of human β-defensin-2, β-defensin-3, and calgranulin by bronchial epithelial cells, essential for killing of bacterial and fungal pathogens.29 A defect in this pathway may explain the recurrent skin and lung infections in hyper-IgE syndrome caused by dominant-negative mutations in STAT3, a transcription factor required for Th17 cell development. Synergistic effects of IL-17 and IL-22 may also play a role in the gut mucosa, on the induction of antimicrobial proteins by gastrointestinal epithelial cells. Hereby these cells, known as Paneth cells, limit dissemination of commensal bacteria that penetrate a disrupted epithelial barrier. These innate and adaptive memory immune cells can be assumed to act in concert to produce a basal level of IL-17 and IL-22, which then maintain a constitutive level of antimicrobial proteins.30 At the same time, phagocytes are generated and attracted to guarantee a first line of defense.
RECOGNITION OF MICROBIAL-DERIVED INFLAMMATORY SIGNALS
Neutrophils bind a wide variety of pathogens and commensal microorganisms, including gram-positive and Gram-negative bacteria, viruses, and fungi. Binding occurs by affinity of neutrophil receptor proteins for common patterns, the pathogen-associated molecular patterns (PAMPs), on the surface of the pathogen. The receptors for PAMPs are called PRRs, and are expressed on the surface of or intracellular in many effector cells of the innate immune system (such as macrophages, dendritic cells, neutrophils) as well as epithelial cells. Whereas immune cells respond vigorously to invading microorganisms, the epithelial linings have to discriminate more selectively to avoid a response to the commensal flora in the guts, the oropharynx, or on our skin. The most common family of PRRs are the Toll-like
receptors (TLRs), which upon ligand binding recruit adaptor molecules to their intracellular signaling domain (FIGURE 47.2).31 This interaction leads to the activation of several kinases that ultimately results in the activation of transcription factor NF-κB and immune-responsive genes and the release of cytokines, for example, TNFα. The TLRs recognize a wide variety of ligands, including lipopeptides by TLR2/TLR1 and TLR2/TLR6 and bacterial lipopolysaccharide (LPS) and flagellin by TLR4 and TLR5. Besides these extracellular pathogen fragments, microbial RNA and DNA are detected by TLR3, 7 and 8, and 9, respectively, all located inside the host cell.
receptors (TLRs), which upon ligand binding recruit adaptor molecules to their intracellular signaling domain (FIGURE 47.2).31 This interaction leads to the activation of several kinases that ultimately results in the activation of transcription factor NF-κB and immune-responsive genes and the release of cytokines, for example, TNFα. The TLRs recognize a wide variety of ligands, including lipopeptides by TLR2/TLR1 and TLR2/TLR6 and bacterial lipopolysaccharide (LPS) and flagellin by TLR4 and TLR5. Besides these extracellular pathogen fragments, microbial RNA and DNA are detected by TLR3, 7 and 8, and 9, respectively, all located inside the host cell.
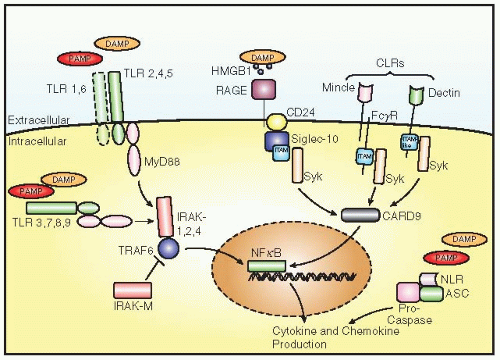 FIGURE 47.2 Pattern recognition receptors (PRRs). Several PRRs recognize pathogen-associated molecular patterns (PAMPs), for example, Toll-like receptors (TLR2/TLR1, TLR2/TLR6, TLR3,-4,-5,-7,-8,-9), C-type lectin receptors (CLRs; Dectin-1,-2, Mincle), and NLRs (NOD-like receptors; NOD1,2). DAMPs bind to carriers like HMGB1 and bind to RAGE as well as to certain TLRs. TLR3 and TLR7 are not expressed on neutrophils. |
Neutrophils express all TLRs, except TLR3 and TLR7.32,33 However, in comparison with monocytes and macrophages, TLR2 and TLR4 are expressed at relatively low levels (in addition to the TLR2/TLR1 and TLR2/TLR6 dimers). This may explain partially why the concentration of LPS required to activate neutrophils is about 100- to 1,000-fold higher than that needed for monocytes.
Most TLR family members have a conserved intracellular signaling motif, the so-called TIR domain. This signaling motif, which is also found in the intracellular domain of the IL-1 receptor (IL-1R), is responsible for NF-κB activation/translocation after TLR or IL-1R ligand binding, and is an essential signaling pathway for IL-1β and TNFα secretion.34 All TLRs signal through MyD88 and Mal, which interact with various IL-1 receptorassociated kinases (IRAKs). However, both TLR3 and TLR4 can also activate cells through a MyD88-independent pathway in which TRIF (TIR-domain-containing adapter-inducing interferon-beta) and TRAM (TRIF-related adaptor molecule) play a role. This can result in phosphorylation and activation of IRF3 or IRF7, which in turn activates a wide range of proinflammatory genes, such as interferon-β. These pathways are specifically active in monocytes and subtypes of dendritic cells. Although mRNA for both TRAM and TRIF is expressed in neutrophils, MyD88-independent signaling in neutrophils does not exist. LPS-responsiveness is strictly IRAK-4 dependent.33
Apart from the TLRs, another family of important surfaceexpressed PRRs is the transmembrane C-type lectin receptors
(CLRs or CLECs).35 The most studied CLR members to date are Dectin-1 (CLEC7a), detecting certain yeast species, and its close homologue Dectin-2 (CLECłn), which recognizes several fungi. All CLRs are activating members of the myeloid-specific Ig superfamily.
The activating members of this family stimulate cells by virtue of cytoplasmic immunoreceptor tyrosine-based activation motifs (ITAMs) in their cytoplasmic tails or in the tails of associated molecules. Tyrosine phosphorylation of these molecules permits binding of phosphotyrosine kinases such as Syk and ZAP-70. Next, tyrosine phosphorylation of these kinases allows binding of tyrosinephosphatases such as SHP1 and 2, which results in activation of NF-κB and the subsequent secretion of cytokines and chemokines. Contrarily, the inhibitory members of the family contain an immunoreceptor tyrosine-based inhibitory motif (ITIM), which has the potency to decrease NF-κB activity.
A third CLR, CLEC4e (also known as Mincle), was also recently shown to recognize fungal pathogens, thereby inducing an intracellular signaling cascade that, besides phosphotyrosine kinases, involves adaptor protein CARD9.36 This ultimately leads to the production of proinflammatory cytokines and chemokines, including TNFα, IL-6, CXCL1 (GROα), and CXCL2 (MIP-2), which stimulate the recruitment of neutrophils to infected tissues.35
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


