During inflammation, leukocytes play a key role in maintaining tissue homeostasis through elimination of pathogens and removal of damaged tissue. Leukocytes migrate to the site of inflammation by crawling over and through the blood vessel wall, into the tissue. Leukocyte adhesion deficiencies (ie, LAD-I, -II, and LAD-I/variant, the latter also known as LAD-III) are caused by defects in the adhesion of leukocytes to the vessel wall, resulting in severe recurrent nonpussing infections and neutrophilia, often preceded by delayed separation of the umbilical cord. Although dependent on the genetic defect, hematopoietic stem cell transplantation is often the only curative treatment.
Key points
- •
During inflammation, leukocytes play a key role in maintaining tissue homeostasis by elimination of pathogens and removal of damaged tissue.
- •
Leukocytes migrate to the site of inflammation by crawling over and through the blood vessel wall, into the tissue.
- •
Leukocyte adhesion deficiencies (ie, LAD-I, -II, and LAD-I/variant, the latter also known as LAD-III) are caused by defects in the adhesion of leukocytes to the blood vessel wall, due to mutations in the genes encoding β2 integrin ( ITGB2 ), a GDP-fucose transport protein ( SLC35C1 ) and kindlin-3 ( FERMT3 ), respectively.
- •
Patients experience recurrent nonpussing bacterial infections and neutrophilia, often preceded by delayed separation of the umbilical cord, and additional symptoms depending on the subtype.
- •
For LAD-I and LAD-III, the only curative treatment is hematopoietic stem cell transplantation. In case of LAD-II, oral fucose supplementation may invert the immune defect, but additional mental retardation is hardly improved.
Introduction
Leukocyte Recruitment and Extravasation
During inflammation, circulating leukocytes migrate to the site of infection following a gradient of chemotaxins in a process called chemotaxis . Chemotaxins may be derived from either the infected tissue or local complement activation, or directly from the pathogens themselves, and diffuse within the tissue into the local vasculature. These gradients of chemotaxins recruit the leukocytes in interplay with factors expressed locally on the luminal side of blood vessel endothelial cells. Neutrophils are short-living leukocytes that are recruited early in the inflammatory response.
Leukocytes following the chemotaxin gradient toward the site of infection must leave the bloodstream, in a process called extravasation ( Fig. 1 ). Extravasation is a multistep process involving adhesion molecules, in which chemotaxins function as activating agents or (pro-) inflammatory mediators. The first step of extravasation consists of initial contact between endothelial cells and leukocytes marginated by the fluid flow of the blood. l -selectin (CD62L) on leukocytes plays a role herein, contacting several cell adhesion molecules on endothelial cells. Within the local environment of an inflammatory tissue reaction, the endothelium begins to express the adhesion molecules p -selectin (CD62P) and later e -selectin (CD62E). The low-avidity interaction of these selectins with their fucosylated ligands on the opposite cells forces the leukocytes to slow down and start a rolling movement along the vessel wall (see Fig. 1 ).
In contrast to the low-avidity binding of leukocytes to selectins, the final step of firm adhesion and subsequent migration depends on stable interaction between integrins on the leukocytes and their ligands on the endothelial cells. Integrins are type 1 transmembrane glycoproteins that form heterodimers via noncovalent association of their α and β subunits, with sizes of 120 to 170 kDa and 90 to 130 kDa, respectively. In mammals, 18 α and 8 β subunits form 24 known combinations, each of which can bind to a specific repertoire of cell-surface, extracellular matrix or soluble protein ligands. The β 2 integrin receptor subfamily is selectively expressed on leukocytes and comprises 4 different heterodimeric proteins, each of which contains a different α subunit: α L β 2 (LFA-1; CD11a/CD18), α M β 2 (CR3; CD11b/CD18), α x β 2 (gp150,95; CD11c/CD18); and α D β 2 (CD11d/CD18), the latter only being expressed on macrophages. The β 2 integrins bind to adhesion molecules on endothelial cells (intercellular adhesion molecule [ICAM]-1 and ICAM-2) and to several complement factors. The main β 2 integrin on neutrophils is CR3.
Slowly rolling leukocytes are able to recognize concentration differences in a gradient of chemotaxins and to direct their movement toward the source of these agents. Although the details of this process remain unknown, the gradient most likely causes a difference in the number of ligand-bound chemotaxin receptors on either side of the cell, thereby inducing the cytoskeletal rearrangements needed for movement. Because adhesion molecules such as the β 2 integrins are essential for the connections with the tissue cells or the extracellular matrix proteins, these connections must be formed at the front of the moving leukocytes and broken at the rear end. Moreover, for continued sensing of the chemotaxin gradient, the chemotaxins must dissociate from their respective receptors for repeated use. This dissociation occurs through internalization of the ligand–receptor complex, intracellular disruption of the connection, and transport of the free receptor to the front of the cell, followed by reappearance of the free receptor on the leukocyte surface. Within the infected tissue, the chemotaxin gradient persists and leukocyte migration is maintained.
Integrin Activation
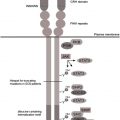
The ligand specificity of integrins is determined by their large extracellular ligand-binding head domain, which is composed of several domains of the α and β subunits. The head domain is attached to the membrane via 2 flexible legs (1 from each subunit), which terminate intracellularly as short cytoplasmic tails. This domain architecture of integrins underlies their ability to transduce bidirectional signals across the plasma membrane: “inside-out” and “outside-in” ( Fig. 2 ). Leukocyte activation (eg, resulting from chemokine binding to chemokine receptors, ligand binding to selectins, or antigen binding to the T-cell receptor) and subsequent intracellular signaling induces conformational changes in the extracellular regions of the β 2 integrins, leading to an enhanced affinity for their ligands (inside-out signaling). In addition, integrins cluster in larger complexes, which increases their ligand avidity. Binding to extracellular ligands leads to further conformational changes of the β 2 integrins, resulting in high ligand affinity, subsequent recruitment of cytosolic proteins, and the initiation of downstream signaling cascades that regulate cell spreading and alter gene expression, cell proliferation, differentiation, and apoptosis (outside-in signaling).
The common activator of most, if not all, integrins is talin, a large cytoskeletal protein that acts as an allosteric activator of integrins by inducing their ligand-binding affinity. The head domain of talin contains a FERM (4.1 protein, ezrin, radixin, moesin) domain, consisting of 3 subdomains: F1, F2, and F3. The F3 subdomain contains a phosphotyrosine-binding (PTB)–like domain that binds to the NPxY/F motif found in the membrane-proximal cytoplasmic region of several β integrins. The head domain is connected to a long cytoplasmic rod that can interact with the cytoskeleton, allowing talin to contribute substantially to adhesion and motility via outside-in signaling cascades.
Although binding of talin seems to be the final step in integrin activation, the activity of talin itself may be regulated by a variety of cell type–specific signaling pathways. A study on chinese hamster ovary (CHO) cells expressing platelet integrins αIIb and β3 proposed that PKCα, activated by phorbol ester phorbol-12-myristate-acetate (PMA) or T-cell receptor stimulation, phosphorylates PKD1, which associates with a small GTPase from the Rap or Ras family that in turn activates the integrin upon formation of an activation complex with its effector protein Rap1-GTP-interacting adaptor molecule (RIAM) and talin.
More recently, kindlin proteins have arisen as key players in integrin activation. Kindlins comprise a family of integrin-binding proteins. In man, the family consists of 3 members: kindlin-1, -2, and -3, which share a high degree of homology. Kindlin-3 is expressed in all hematopoietic cell types, in which it plays an important role in a variety of functions depending on integrin-mediated adhesion, such as platelet clot formation and leukocyte extravasation. Biochemical studies have confirmed that all kindlins directly bind synthetically generated cytoplasmic tails of β 1 , β 2 , and β 3 integrins. Because kindlins possess a FERM domain that is homologous to that of talin, it was hypothesized that kindlins, like talin, interact with the membrane-proximal NPxY/F motif in the cytoplasmic tail of β integrins. However, recent studies have shown that the kindlin-binding site of β integrins is distinct from the talin-binding site, ie, at a membrane-distal NxxY/F motif in the cytoplasmic integrin tail. Biochemical studies with mutants of kindlin-2 have shown that the PTB domain in F3 is, similarly to talin, essential for integrin binding, in addition to the N -terminus of the protein being required for interaction with β 3 . Kindlins are essential for inside-out integrin activation, and recent reports indicate an additional role in downstream outside-in cascades.
Leukocyte Adhesion Deficiency
Leukocyte adhesion deficiencies (ie, LADs I, II, and III, the latter also known as LAD-1/variant) are immunodeficiencies caused by defects in the adhesion of leukocytes (especially neutrophils) to the blood vessel wall. As a result, patients with any LAD subtype experience severe bacterial infections and neutrophilia, often preceded by delayed separation of the umbilical cord ( Table 1 ). LAD-II is characterized by additional developmental problems, whereas in LAD-III, the immune defects are supplemented by a Glanzmann thrombasthenia-like bleeding tendency.
| LAD-I | LAD-II | LAD-III | |
|---|---|---|---|
| Physical examination | |||
| Recurrent nonpurulent bacterial and fungal infections | ++ (skin & mucosal, gingivitis, periodontitis; necrotizing) | + (decreasing during childhood; periodontitis) | + (all have bacterial, some have fungal) |
| Delayed separation of umbilical cord, wound healing defect | + (common omphalitis) | – | + (some omphalitis) |
| Dysmorphism | – | + (growth retardation, short stature, coarse face) | – |
| Neural abnormalities | – | + (mental retardation, autism, convulsions, cerebral atrophy) | – |
| Bleeding tendency | – | – | + |
| Imaging and additional testing | |||
| Genetic defect | + ( ITGB2 ) | + ( SLC35C1 ) | + ( FERMT3 ) |
| Leukocytosis | + (20.10 6 up to 140.10 6 /mL) | + (up to 150.10 6 /mL during infections) | + (20.10 6 up to 70.10 6 /mL, of which 30%–90% are neutrophils) |
| Erythrocytosis | – | – | Hb, 6–9/12–14 |
| Thrombocytosis | – | – | Plt, 20–320 (avg, 140) |
| Expression of integrins (CD18, CD11b) | 1%–30%, or present but dysfunctional | + | + |
| Expression of fucosylated glycoconjugates | + | – (sLeX, LeX, H-antigen, N -glycans) | + |
| Neutrophil defects | + (firm adhesion, chemotaxis) | + (rolling) | + (firm adhesion, chemotaxis, spreading) |
| Platelet defects | – | – | + (aggregation) |
| Therapeutic options | |||
| Antibiotics | + | + | + (also prophylactic) |
| HSCT | + (severe vs moderate) | ? | + |
| Fucose supplementation | – | + | – |
| Granulocyte transfusion | + (potential benefit established) | + (potential benefit established) | |
| Thrombocyte transfusion | _ | _ | ++/– (varies per patient) |
| Survival without HSCT | Severe form: low Moderate form: better | High | Low (10%–20%) often prior siblings diseased |
Introduction
Leukocyte Recruitment and Extravasation
During inflammation, circulating leukocytes migrate to the site of infection following a gradient of chemotaxins in a process called chemotaxis . Chemotaxins may be derived from either the infected tissue or local complement activation, or directly from the pathogens themselves, and diffuse within the tissue into the local vasculature. These gradients of chemotaxins recruit the leukocytes in interplay with factors expressed locally on the luminal side of blood vessel endothelial cells. Neutrophils are short-living leukocytes that are recruited early in the inflammatory response.
Leukocytes following the chemotaxin gradient toward the site of infection must leave the bloodstream, in a process called extravasation ( Fig. 1 ). Extravasation is a multistep process involving adhesion molecules, in which chemotaxins function as activating agents or (pro-) inflammatory mediators. The first step of extravasation consists of initial contact between endothelial cells and leukocytes marginated by the fluid flow of the blood. l -selectin (CD62L) on leukocytes plays a role herein, contacting several cell adhesion molecules on endothelial cells. Within the local environment of an inflammatory tissue reaction, the endothelium begins to express the adhesion molecules p -selectin (CD62P) and later e -selectin (CD62E). The low-avidity interaction of these selectins with their fucosylated ligands on the opposite cells forces the leukocytes to slow down and start a rolling movement along the vessel wall (see Fig. 1 ).
In contrast to the low-avidity binding of leukocytes to selectins, the final step of firm adhesion and subsequent migration depends on stable interaction between integrins on the leukocytes and their ligands on the endothelial cells. Integrins are type 1 transmembrane glycoproteins that form heterodimers via noncovalent association of their α and β subunits, with sizes of 120 to 170 kDa and 90 to 130 kDa, respectively. In mammals, 18 α and 8 β subunits form 24 known combinations, each of which can bind to a specific repertoire of cell-surface, extracellular matrix or soluble protein ligands. The β 2 integrin receptor subfamily is selectively expressed on leukocytes and comprises 4 different heterodimeric proteins, each of which contains a different α subunit: α L β 2 (LFA-1; CD11a/CD18), α M β 2 (CR3; CD11b/CD18), α x β 2 (gp150,95; CD11c/CD18); and α D β 2 (CD11d/CD18), the latter only being expressed on macrophages. The β 2 integrins bind to adhesion molecules on endothelial cells (intercellular adhesion molecule [ICAM]-1 and ICAM-2) and to several complement factors. The main β 2 integrin on neutrophils is CR3.
Slowly rolling leukocytes are able to recognize concentration differences in a gradient of chemotaxins and to direct their movement toward the source of these agents. Although the details of this process remain unknown, the gradient most likely causes a difference in the number of ligand-bound chemotaxin receptors on either side of the cell, thereby inducing the cytoskeletal rearrangements needed for movement. Because adhesion molecules such as the β 2 integrins are essential for the connections with the tissue cells or the extracellular matrix proteins, these connections must be formed at the front of the moving leukocytes and broken at the rear end. Moreover, for continued sensing of the chemotaxin gradient, the chemotaxins must dissociate from their respective receptors for repeated use. This dissociation occurs through internalization of the ligand–receptor complex, intracellular disruption of the connection, and transport of the free receptor to the front of the cell, followed by reappearance of the free receptor on the leukocyte surface. Within the infected tissue, the chemotaxin gradient persists and leukocyte migration is maintained.
Integrin Activation
The ligand specificity of integrins is determined by their large extracellular ligand-binding head domain, which is composed of several domains of the α and β subunits. The head domain is attached to the membrane via 2 flexible legs (1 from each subunit), which terminate intracellularly as short cytoplasmic tails. This domain architecture of integrins underlies their ability to transduce bidirectional signals across the plasma membrane: “inside-out” and “outside-in” ( Fig. 2 ). Leukocyte activation (eg, resulting from chemokine binding to chemokine receptors, ligand binding to selectins, or antigen binding to the T-cell receptor) and subsequent intracellular signaling induces conformational changes in the extracellular regions of the β 2 integrins, leading to an enhanced affinity for their ligands (inside-out signaling). In addition, integrins cluster in larger complexes, which increases their ligand avidity. Binding to extracellular ligands leads to further conformational changes of the β 2 integrins, resulting in high ligand affinity, subsequent recruitment of cytosolic proteins, and the initiation of downstream signaling cascades that regulate cell spreading and alter gene expression, cell proliferation, differentiation, and apoptosis (outside-in signaling).
The common activator of most, if not all, integrins is talin, a large cytoskeletal protein that acts as an allosteric activator of integrins by inducing their ligand-binding affinity. The head domain of talin contains a FERM (4.1 protein, ezrin, radixin, moesin) domain, consisting of 3 subdomains: F1, F2, and F3. The F3 subdomain contains a phosphotyrosine-binding (PTB)–like domain that binds to the NPxY/F motif found in the membrane-proximal cytoplasmic region of several β integrins. The head domain is connected to a long cytoplasmic rod that can interact with the cytoskeleton, allowing talin to contribute substantially to adhesion and motility via outside-in signaling cascades.
Although binding of talin seems to be the final step in integrin activation, the activity of talin itself may be regulated by a variety of cell type–specific signaling pathways. A study on chinese hamster ovary (CHO) cells expressing platelet integrins αIIb and β3 proposed that PKCα, activated by phorbol ester phorbol-12-myristate-acetate (PMA) or T-cell receptor stimulation, phosphorylates PKD1, which associates with a small GTPase from the Rap or Ras family that in turn activates the integrin upon formation of an activation complex with its effector protein Rap1-GTP-interacting adaptor molecule (RIAM) and talin.
More recently, kindlin proteins have arisen as key players in integrin activation. Kindlins comprise a family of integrin-binding proteins. In man, the family consists of 3 members: kindlin-1, -2, and -3, which share a high degree of homology. Kindlin-3 is expressed in all hematopoietic cell types, in which it plays an important role in a variety of functions depending on integrin-mediated adhesion, such as platelet clot formation and leukocyte extravasation. Biochemical studies have confirmed that all kindlins directly bind synthetically generated cytoplasmic tails of β 1 , β 2 , and β 3 integrins. Because kindlins possess a FERM domain that is homologous to that of talin, it was hypothesized that kindlins, like talin, interact with the membrane-proximal NPxY/F motif in the cytoplasmic tail of β integrins. However, recent studies have shown that the kindlin-binding site of β integrins is distinct from the talin-binding site, ie, at a membrane-distal NxxY/F motif in the cytoplasmic integrin tail. Biochemical studies with mutants of kindlin-2 have shown that the PTB domain in F3 is, similarly to talin, essential for integrin binding, in addition to the N -terminus of the protein being required for interaction with β 3 . Kindlins are essential for inside-out integrin activation, and recent reports indicate an additional role in downstream outside-in cascades.
Leukocyte Adhesion Deficiency
Leukocyte adhesion deficiencies (ie, LADs I, II, and III, the latter also known as LAD-1/variant) are immunodeficiencies caused by defects in the adhesion of leukocytes (especially neutrophils) to the blood vessel wall. As a result, patients with any LAD subtype experience severe bacterial infections and neutrophilia, often preceded by delayed separation of the umbilical cord ( Table 1 ). LAD-II is characterized by additional developmental problems, whereas in LAD-III, the immune defects are supplemented by a Glanzmann thrombasthenia-like bleeding tendency.
Related posts:
 Genetics and Pathophysiology of Severe Congenital Neutropenia Syndromes Unrelated to Neutrophil Elastase
Defective G-CSFR Signaling Pathways in Congenital Neutropenia
Genetics and Pathophysiology of Severe Congenital Neutropenia Syndromes Unrelated to Neutrophil Elastase
Defective G-CSFR Signaling Pathways in Congenital Neutropenia
 Granulocyte Colony-Stimulating Factor Receptor Signaling
Genetics and Pathophysiology of Severe Congenital Neutropenia Syndromes Unrelated to Neutrophil Elastase
Granulocyte Colony-Stimulating Factor Receptor Signaling
Defective G-CSFR Signaling Pathways in Congenital Neutropenia
Granulocyte Colony-Stimulating Factor Receptor Signaling
Genetics and Pathophysiology of Severe Congenital Neutropenia Syndromes Unrelated to Neutrophil Elastase
Granulocyte Colony-Stimulating Factor Receptor Signaling
Defective G-CSFR Signaling Pathways in Congenital Neutropenia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


