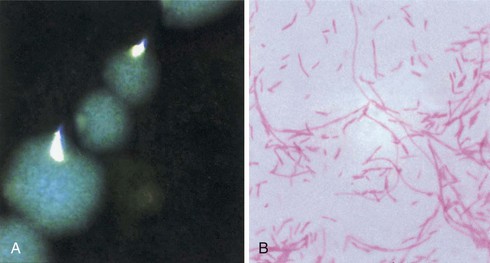
Paul H. Edelstein, Craig R. Roy Keywords air conditioning; antigen detection; community-acquired pneumonia; cooling towers; epidemic; hot tub–associated infections; hot water plumbing; hyponatremia; immunosuppressed patients; infectious aerosols; legionnaires’ disease; Legionella; longbeachae; macrophages; micdadei; nosocomial; opportunistic infections; pneumonia; pneumophila; waterborne infections Legionnaires’ disease (LD) is an acute pneumonic illness caused by gram-negative bacilli of the genus Legionella, the most common of which is Legionella pneumophila (Lp). Pontiac fever is a febrile, nonpneumonic, systemic illness closely associated with, if not caused by, Legionella spp. Legionellosis is the term that encompasses all diseases caused by, or presumed to be caused by, the Legionella bacteria, including LD, focal nonpulmonary infections, and Pontiac fever. LD was first recognized when it caused an epidemic of pneumonia at a Pennsylvania State American Legion convention in Philadelphia in 1976; 221 people were affected, and 34 died. Despite intensive laboratory investigation, the cause of the outbreak went undetected for many months. Epidemiologic investigation concluded that the disease was most likely airborne and focused primarily at one convention hotel, which closed because of adverse publicity.1,2 About 6 months later, Joseph McDade and Charles Shepard, investigators at the Centers for Disease Control and Prevention (CDC), announced that they had discovered the etiologic agent, a fastidious gram-negative bacillus.3 Because of the historical association with the American Legion convention, this disease is now called “Legionnaires’ disease,” and the etiologic agents belong to the family Legionellaceae, with Lp being the agent responsible for the 1976 Philadelphia epidemic. Antibody testing for the disease showed that several prior unsolved outbreaks of pneumonia had been LD, including epidemics investigated in the 1950s and 1960s.4–6 An unsolved epidemic of a nonpneumonic febrile illness in Pontiac, Michigan, was also found to have been due to exposure to Legionella bacteria; this illness was termed “Pontiac fever.”7,8 Prior epidemics of Pontiac fever occurred as early as 1949, without determination of an etiology.9 Bacterial culture isolates from the 1940s through the 1960s were found to be Legionella bacteria, although at the time they had been thought to be rickettsial agents.10–15 Thus both the organism and the disease had been studied decades before, but major advances in technology and epidemiology were required to properly classify the disease and determine its cause. Even with identification of Lp in 1977 as the cause of LD, the source of the bacterium, factors promoting its multiplication and spread, and ways to abort epidemics of LD remained uncertain for several years. Epidemics of the disease, especially nosocomial ones, commonly lasted for years.16–22 Eventually it was discovered that Lp and other Legionella spp. were naturally occurring aquatic bacteria that had a propensity for growing in warm water, most particularly in cooling towers, water heaters, and potable water plumbing. These discoveries led to the end of several multiyear outbreaks of the disease and to methods for preventing the disease.23 It is now unusual for LD outbreaks to last more than 1 or 2 weeks. LD still occurs, both in sporadic and epidemic form, sometimes involving many hundreds of victims.24–26 The disease, although a relatively rare (1% to 5%) cause of community-acquired pneumonia, can cause severe disease if treated improperly. Enough is now known about the disease so that epidemics can be aborted in days, to effectively treat affected patients, and to reduce the frequency of the disease by making modifications in building design and maintenance. The Legionella spp. are small gram-negative bacilli with fastidious growth requirements. Proteins rather than carbohydrates are used as an energy source. Obligate aerobes, the bacteria grow at temperatures ranging from 20° C to 42° C. Coxiella burnetii, an obligate intracellular parasite and the etiologic agent of Q fever, is the closest relative of the Legionellaceae. L-cysteine is required for the growth of all but one of the clinically important Legionella spp., and this amino acid is necessary for the initial growth of all described Legionella spp. from environmental or clinical sources. Soluble iron is required for optimal growth and for initial isolation of the bacterium from both clinical and environmental sources. Iron, L-cysteine, α-ketoglutarate, and charcoal-containing yeast extract agar buffered with an organic buffer (BCYEα agar) create the preferred growth medium for clinical isolation. Clinically important Legionella spp. grow best at 35° C in humidified air on BCYEα medium, usually in 2 to 5 days after inoculation of plates. Up to 14 days’ incubation may rarely be required for the isolation of unusual Legionella spp. More than 58 different Legionella spp. have been described, about half of which have been reported to infect humans.27,28 Lp contains at least 16 different serogroups; seven other species contain two different serogroups, with the remaining species containing only one serogroup each.29 Lp serogroup 1 caused the 1976 Philadelphia outbreak and is the cause of 65% to 90% of all cases of LD for which there is a bacterial isolate.30–32 Lp serogroup 1 dominance is not universal. For example, Lp serogroup 1 constitutes only 66% of Legionella spp. isolates in LD patients in Ontario Province, Canada, with Lp serogroup 6 being a relatively common isolate in that province.32 Also, L. longbeachae (Llb) causes 30% to 50% of LD cases in New Zealand and a similar fraction in Australia.33 Lp serogroup 1 can be divided into multiple subtypes using a variety of serologic, other phenotypic, and genetic methods. One particular subtype of Lp serogroup 1 causes 55% to 76% of cases of LD due to Lp and 85% of cases due to Lp serogroup 1; this subtype is distinguished by its reactivity with a particular monoclonal antibody and is termed the “Pontiac” subtype.34,35 The predominance of the Pontiac subtype has implications for diagnosis. The most common non-Lp species that are isolated from humans are Llb, L. micdadei, L. bozemanae, and L. dumoffii,31 which with the exception of Australasia, constitute less than 5% of culture-proven cases.36,37 Most clinical microbiology laboratories should be able to identify Legionella bacteria to the genus level by detection of their typical colony morphology and Gram stain appearance, by determination of L-cysteine growth dependence, and by excluding possible mimics by using standard microbiology identification techniques (Fig. 234-1). Identification of Lp and Lp serogroup 1, the most common clinical isolate, can be accomplished by sophisticated clinical microbiology laboratories using relatively simple serologic testing. Identification of other Lp serogroups and other Legionella spp. is often much more difficult and requires molecular testing for definitive identification.38 Legionella bacteria are found in our natural aqueous environment, in lakes, streams, and even coastal oceans, at temperatures ranging from 5° C to greater than 50° C.39 Warm water (25° C to 40° C) supports the highest concentration of these bacteria, with warm water being the major bacterial reservoir leading to LD. Free-living amebas in the same waters support the intracellular growth and survival of Legionella bacteria. The interaction between amebas and Legionella bacteria has been studied most comprehensively for Lp, which is a facultative intracellular parasite of several different amebas.40,41 The bacterium multiplies many thousand-fold within the amebas (Fig. 234-2). When faced with inimical environmental factors, such as pH changes, absence of nutrients, or temperature alteration, the Legionella-infected amebas encyst, guaranteeing the survival of both the host and parasite until more favorable conditions allow excystment. In both natural and manmade waters, Legionella-infected amebas are found in consortia of many different microorganisms, all of which exist in a biofilm. In addition to intra-amebal survival, freely living Legionella bacteria can enter a low metabolic state termed “viable but not cultivatable,” making them difficult to recover from the environment and biocide-resistant.42 The Legionella bacteria, amebas, and other microorganisms constantly escape from the biofilm (“sessile phase”) because of water flow and pressure fluxes into a freely moving phase (“planktonic phase”) and then back into the sessile phase. Environmental changes that disrupt the biofilm can result in the sudden and massive release of Legionella bacteria into the surrounding water. If this water is then aerosolized or aspirated, the bacteria can cause illness in a susceptible host. Almost all cases of LD result from Legionella contamination of warm manmade water sources, such as water heaters, air conditioning and other types of cooling towers, warm water baths, warm water plumbing systems, and recirculating water systems. Some recently reported novel water sources include rain puddles in tropical regions, tsunami-related water exposure, windshield wiper fluid, and water from truck tankers used to clean or maintain roads.43–45 One exception to water as a risk factor is that Llb appears to be transmitted mainly through soil contact, especially potting soil used by gardeners.37 Legionella bacteria are present in low concentrations in disinfectant-treated cold potable water, usually at levels of less than one bacterium per liter; up to 50 L of such water may need to be sampled to detect a single Legionella bacterium. However, within water distribution pipes, especially older pipes with low or no water flow, the bacterial density can be amplified by growth in biofilm. The bacteria can be further amplified in the presence of warm conditions, such as those found in many buildings or heat rejection devices. Legionella bacteria concentrations in air conditioning cooling towers range from 102 to 108 colony-forming units (CFU)/L. Up to 80% of air conditioning cooling towers tested contain the bacterium, as do 5% to 30% of home and industrial water heaters and hot water plumbing.39 Contaminated water that is then aerosolized serves as a disseminator of the bacteria into the environment. The concentration of Legionella bacteria in a particular environmental site may spontaneously fluctuate over a wide range due to changes in temperature, the presence of biofilm disruptive forces, the type and concentration of other microorganisms in the biofilm consortium, and the concentrations of a variety of organic and inorganic compounds. LD is initiated by inhalation, and probably microaspiration, of Legionella bacteria into the lungs. Although Legionella bacteria are ubiquitous in our environment, they rarely cause disease. A confluence of a number of factors must occur simultaneously before LD is possible. These factors include the presence of virulent strains in an environmental site; a means for dissemination of the bacteria, such as by aerosolization; and proper environmental conditions allowing the survival and inhalation of an infectious dose of the bacteria by a susceptible host. Strains of different virulence exist for the same species, and some species and serogroups are more virulent than others.46–48 Possible strain virulence factors include aerosol stability, ability to grow within macrophages, possession of eukaryotic gene homologues, and surface hydrophobicity. The infectious form of the bacterium is not known, but in all cases the bacteria originate from water or soil. Several possibilities exist for the infectious particle causing disease, including Legionella bacteria contained within an amebal cyst, a sporelike form of the bacterium,49 a particle of biofilm containing Legionella bacteria and other microorganisms, and freely dispersed extracellular planktonic Legionella bacteria. The physiologic state of Legionella bacteria may be important for its virulence because virulence increases when the bacterium is grown in amebas, in the late stationary phase in vitro, or as the sporelike form.50,51,52 The bacterial inoculum required to cause LD is unknown. Guinea pigs develop asymptomatic infection with inocula as low as 10 to 100 bacteria when delivered by aerosol, disease with approximately 1000 bacteria, and death after infection with 10,000 bacteria.53,54 A packet of bacteria in amebal cysts or in a biofilm fragment easily contains more than 1000 bacteria, making it possible that inhalation of a single infected amebal cyst or biofilm fragment could cause disease.55 Survival of aerosolized extracellular Lp is dependent on relative humidity, with strain-dependent humidity optima, meaning that the disease-causing critical concentration of bacteria in the environment may differ even for the same strain, depending on environmental conditions.56 Relative humidity may be a key factor in disease transmission.25,57 After bacteria enter the lung they are phagocytosed by alveolar macrophages. The Legionella bacteria produce virulence factors that enhance phagocytosis and promote intracellular survival and replication (see later). After sufficient intracellular replication, the bacteria kill the macrophage, escape into the extracellular environment, and are then rephagocytosed by macrophages. The bacterial concentration in the lung increases due to amplification of the bacteria within macrophages; the Lp concentration in guinea pig lungs increases by about 1 million-fold over a 3-day period after initial infection. Following this intracellular multiplication, neutrophils, additional macrophages, and erythrocytes infiltrate the alveoli and capillary leakage results in edema.58 Chemokines and cytokines released by infected macrophages help trigger the severe inflammatory response.59 In the A/J mouse model of LD,60 the relevant proinflammatory chemokines and cytokines include KC, MIP-2, tumor necrosis factor (TNF)-α, interleukin (IL)-12, IL-18, and interferon-γ (IFN-γ).61–63 Humans with LD had elevated levels of TNF-α and IL-8 in relation to other bacterial pneumonias in one study.64 Systemic spread of the bacteria may be accomplished by infection of circulating monocytes. The mechanisms for systemic toxicity of the disease are unclear, but it involves the severe inflammatory response to virulent Lp. Cytokine production is mediated by detection of microbial products by receptors of the innate immune system. Toll-like receptors (TLRs) on macrophages and other cells initiate host responses to both virulent Lp and avirulent mutants by detecting common microbial-associated molecular patterns (MAMPs). TLR2 detects lipoproteins and lipopeptides; TLR 5, flagellin; and TLR9, bacterial DNA.65–67 Replication of Lp is greater in the lungs of mice deficient for TLR2 or the TLR adapter protein MyD88, resulting in higher mortality,68,69 and mice lacking TLR5 or TLR9 have delayed innate immune responses but disease susceptibility is not as pronounced as in TLR2-deficient mice.67,70,71 Epidemiologic data indicate that humans with a TLR5 stop polymorphism are more susceptible to LD; for unclear reasons human TLR4 polymorphisms are protective,65–67,72 even though the lipid A moiety produced by Lp has an atypical structure and is less stimulatory toward TLR4 than the classic lipid A molecule produced by enterobacteria.73 Other eukaryotic cell surface receptors may be important, as demonstrated by the association of mannose-binding lectin deficiency with human susceptibility to LD.74 The delivery of microbial products into the macrophage cytosol by virulent Lp contributes significantly to the robust inflammatory response.75 Many responses are activated by host nucleotide-binding, oligomerization domain (NOD)–like receptor (NLR) proteins. The mouse NLR protein NAIP5 detects bacterial flagellin to activate caspase-1-dependent processing and secretion of the cytokines IL-1β and IL-18,76–78 and animals that are defective for NAIP5 are significantly more susceptible to Lp infection.79,80 The NLR proteins NOD1 and NOD2 respond to peptidoglycan fragments delivered into the cytosol by virulent Lp and sustain activation of innate immune signaling pathways.81,82 Lastly, the proteins RIG-I and MAVS respond to bacterial nucleic acid delivered into the cytosol during Lp infection to activate type I interferon production.83,84 Although the severe Th1 response induced by Lp can be detrimental to the host under high bacterial loads, these cytokines are crucial for the clearance of Legionella organisms.62 Cytokines produced by infected alveolar macrophages are essential for the recruitment of neutrophils and for stimulating IFN-γ production by natural killer (NK) cells, which are both needed for sterilization in the lung.85 Neutrophils are efficient at clearing extracellular Lp from the lungs. Macrophages activated by IFN-γ kill Lp.86 This change in macrophage permissiveness involves, among other things, a reduction in intracellular iron, a factor that is necessary for Lp replication.87 Indeed, the majority of legionellae seen in lung samples are associated with alveolar macrophages. Furthermore, the susceptibility of an animal species correlates with the ability of Lp to infect its macrophages, and bacterial mutants that are impaired for in vitro infection of macrophages have reduced virulence. Antibodies develop during the course of Lp infection, but the humoral immune response does not appear to be critical for host defense. It is widely believed that the adaptation of Lp to protozoan niches in nature engendered it with the ability to infect mammalian phagocytes.88 Lp enters the macrophage by conventional or coiling phagocytosis,89–91 processes that utilize the host cell actin cytoskeleton.92 Opsonization with the C3 component of complement can promote phagocytosis,93 but entry by this pathway dampens the oxidative burst and thereby may enhance bacterial intracellular survival. However, opsonin-independent phagocytosis also appears to be important.52 Even in the event that the oxidative burst is triggered, Lp strains may be resistant to hydrogen peroxide, superoxide anion, and hydroxyl radicals. After entry, legionellae reside within a nascent phagosome (Fig. 234-3) that does not fuse with endosomes or lysosomes,94–96 thereby avoiding acidification and degradative enzymes. The phagosome subverts host vesicles in the early secretory pathway using proteins produced by Lp.97 The vacuole containing Lp rapidly recruits vesicles as they exit the endoplasmic reticulum and develops into an organelle that resembles the host rough endoplasmic reticulum.98–100 This specialized Legionella-containing vacuole (LCV) supports intracellular replication. This vacuole expands during replication and ultimately fills the host cell. On nutrient depletion (e.g., amino acid depletion), Lp enters a stationary phase and converts to a flagellated form that is primed to seek out and infect new host cells.50,101 Egress of Lp from the expended host cell is not well understood, but in macrophages this involves pathogen-induced apoptosis that leads to cellular necrosis.102,103 Processes in addition to macrophage infection likely contribute to disease caused by Lp. The bacterium may replicate or, at a minimum, must temporarily survive within the extracellular spaces of the alveoli.104 The ability of Lp strains to resist complement and cationic peptides may be especially relevant for extracellular survival, particularly following the onset of inflammation.105–107 The examination of infected lung tissue suggests that Lp may also grow within the alveolar epithelium.108 On the basis of in vitro models, the microbe grows within both alveolar type I and type II cells.109,110 The importance of extramacrophage processes is also implied by the fact that mutants can be isolated that are not defective for macrophage infection but are impaired for virulence in animals.111 Neither the pathogenesis nor the etiology of Pontiac fever is known with certainty. Pontiac fever is caused by inhalation of a disease-causing environmental aerosol derived from water containing microorganisms including Legionella bacteria. Thirty to 85% of patients with this disease have serum anti-Legionella antibody in higher concentrations than is found in the normally healthy population.112 The prevalent assumption is that the illness is caused by inhalation of the Legionella bacteria. However, because the aerosols contain a vast array of other microorganisms and toxins, including endotoxin, it is unclear whether the disease is due to inhalation of endotoxin, to inhalation of a number of microorganisms, to inhalation of Legionella bacteria alone, or to a combination of all these agents.112 The short incubation period, 4 to 6 hours, of some Pontiac fever patients supports a toxin-mediated illness, while the median incubation period of around 35 hours is more suggestive of initial bacterial multiplication causing illness. Infections with non-Legionella bacteria can produce Legionella antibodies, so the presence of such antibodies does not prove that Pontiac fever is due solely to Legionella infection or intoxication. Bath water fever, a clinical syndrome thought to be due to endotoxin inhalation, is similar to Pontiac fever, suggesting that Pontiac fever may also be caused by endotoxin inhalation.112 Several reports exist in which exposure to the same environmental source led to Pontiac fever in most exposed people, but to LD in a few people; whether the milder illness was really Pontiac fever or mild LD is open to question and does not help answer the question of etiology and pathogenesis.112 Perhaps the strongest evidence implicating systemic infection with Legionella bacteria as the cause of Pontiac fever has been the rare reports of positive Lp urinary antigen tests or positive cultures in patients with Pontiac fever.112 The rarity of such cases and the rapid recovery without antibiotic therapy argue against systemic infection as the cause of Pontiac fever. A variety of surface structures have been implicated in Lp pathogenesis. Type IV pili modestly promote bacterial attachment to macrophages and epithelial cells,113 and flagella promote invasion independent of adherence.114 The major outer membrane protein is a porin that also serves as a binding site for complement components and thus mediates opsonophagocytosis.115 The Mip protein is a surface-exposed peptidyl prolyl isomerase that is required for the early stages of intracellular infection and for full virulence in animals,116–118 whereas the heat shock protein Hsp60 has been shown to enhance epithelial cell invasion.119 The rtxA gene promotes adherence and virulence, although the structure and localization of its protein product are unclear.120,121 Legionella LPS contains some endotoxic activity, and changes in LPS have correlated with increases in serum resistance, intracellular growth, and virulence.122 Finally, the rcp gene, which appears to encode a lipid A modifying enzyme, confers resistance to cationic peptides and promotes macrophage and lung infection.106 Lp secretes a variety of proteins, degradative enzymes, and putative toxins. The release of proteins by Lp into the extracellular milieu is mediated primarily by a type II secretion system.81,123–125 Genome sequencing also suggests the existence of type I and type V secretion systems.126 Acid phosphatases; aminopeptidases; an RNase; a chitinase; a zinc metalloprotease; monoglycerol, diglycerol, and triacylglycerol lipases; a phospholipase A; a lysophospholipase A; cholesterol acyltransferase; phospholipases C; a peptidyl prolyl isomerase; and a number of novel proteins are all secreted via the Legionella type II system.127–130 Mutations within the genes encoding the type II secretion system diminish infectivity for macrophages, protozoa, and animals.129,131 The secreted zinc protease is produced during infection and promotes pathology in the guinea pig model of disease, as well as intracellular infection of some ameba hosts.127,132 Surprisingly, the type II-secreted chitinase promotes bacterial persistence in the lungs of infected mice.128 A Legionella type IV secretion system called Dot/Icm is essential for intracellular replication and virulence in animal models of disease.81 First, it enhances Lp entry into host cells.133,134 Second, it is essential for the ability of the Legionella parasite to modulate host vesicular transport to avoid delivery to lysosomes and promote vacuole remodeling by the host vesicles in the early secretory pathway.95,135,136 Finally, the Dot/Icm system is important for apoptosis and bacterial egress from its spent host cell.137,138 Mutations in dot/icm loci lead to loss of virulence.111,139 Many hundreds of proteins are secreted by the Dot/Icm system, and in most cases these “effector” proteins translocate from the Legionella-containing vacuole into the host cell cytoplasm.81,140 Some of these, including VipA, VipD, VipF, AnkX, LegC3, LegC7 (YflA), and LegC2 (YlfB), have been implicated in bacterial evasion of lysosome fusion.141–143 Others, such as RalF, DrrA (SidM), LepB, LidA, and SidJ, play roles in the recruitment of endoplasmic reticulum–derived vesicles to the Legionella-containing vacuole.144–147 The effector SdhA is important for maintaining the integrity of the vacuole in which Lp resides,148 and SidF is a lipid phosphatase that generates phosphatidylinositol phosphate signatures on the cytosolic surface of the vacuole that are important for the binding of other effectors.149 The effectors LepA and LepB have been implicated in bacterial egress from the spent host cell.150 Lp expresses several effectors that function as glucosyltransferases that are capable of inhibiting host cell protein synthesis (elongation factor 1A).151,152 Although Dot/Icm is generally regarded as essential for Lp infection, a second type IV secretion system known as Lvh is found in addition to Dot/Icm in many strains; however, this system does not appear to deliver effector proteins into host cells but retains the ability to promote DNA conjugation, which could facilitate horizontal transfer of genes encoding effectors.153 Interestingly, many of the type II– and type IV–secreted proteins, as well as other mediators of infection (e.g., LpnE, Lpg0971), bear striking sequence similarity to eukaryotic proteins, suggesting that they were acquired by horizontal gene transfer from a eukaryotic host and use similar biochemical activities to facilitate Lp infection.126,128,141,154–158 Several infectivity factors have been localized to the Lp periplasm or cytoplasm. A Cu-Zn superoxide dismutase resides in the periplasm, affording resistance to toxic superoxide anions, and the KatB catalase-peroxidase is necessary for optimal intracellular infection.159,160 The Legionella phosphoenolpyruvate phosphotransferase and HtrA protein promote intracellular growth and virulence.161,162 Lp iron acquisition, important for intracellular and extracellular replication, involves, among other things, a secreted ferric iron chelator (the siderophore legiobactin), a secreted pyomelanin with ferric reductase activity, and an inner membrane ferrous iron transporter (FeoB).163–166 In addition to the identification of eukaryotic-like proteins in the Lp genome, another outcome of sequencing bacterial genomes is the realization that there are large segments of DNA, including plasmids and chromosomal “islands,” which can vary between Lp strains.126,167–169 It is possible that these variable regions of the genome will help explain differences in virulence that may exist between strains. Indeed, large deletions of the chromosome that remove individual islands can decrease Lp replication in specific protozoan hosts.170 Relatively little is known about the virulence mechanisms, molecular pathogenesis, and cell biology of infections caused by Legionella spp. other than Lp. With the major exception of Llb, infections caused by the other Legionella spp. are rare and found almost exclusively in severely immunocompromised patients. This implies that these other species lack critical virulence factors that allow ready intracellular multiplication. However, L. micdadei, L. dumoffii, Llb, and other species can multiply normally in guinea pig and other animal macrophages.171–174 Unlike Lp, non-AJ strain mice are not resistant to infection with a number of other Legionella spp., including L. bozemanae, L. dumoffii, L. micdadei, and Llb.175 Despite unrestricted growth in otherwise unpermissive macrophages, some non-Lp species fail to promote inflammatory cell death,176 which could be correlated to a failure to cause disease in nonimmunocompromised people, although this is unstudied. Llb resides within a ribosome-studded phagosome, albeit with markers of late endosomal maturation, unlike Lp, which can block endosomal maturation at an early stage.171,177,178 In contrast, L. micdadei resides in a smooth phagosome, and morphologic data suggest L. dumoffii grows within the cytoplasm rather than in a phagosome.174,177,179,180 Analysis of the genome of several different Llb strains has shown that this species has and utilizes the Dot/Icm system for causing infection, that it is nonflagellated, and that it possesses a wide range of eukaryotic effectors different from those found in Lp.181 Many of the Llb effectors appear to have been derived from plants and soil organisms, suggesting that it has evolved as primarily a soil-adapted bacterium. Experimental virulence of Llb for macrophages appears to be relatively independent of growth phase in contrast to Lp.171,182 The incubation period during most outbreaks of LD is between 2 and 10 days, with median values of 4 to 6 days, and some outliers from 1 up to 28 days.1,183 A 2-month incubation period was reported for one nosocomial case.184 The incubation period of Pontiac fever is from 4 hours to 3 days, with a median of around 32 to 36 hours, although incubation periods of up to 5 days have been reported.7,185,186 The incubation period of LD due to Legionella species other than Lp is not known with certainty; on the basis of case reports from nosocomial cases of the disease, the incubation period after onset of immunosuppression appears to be similar to that of LD due to Lp.187 Person-to-person transmission of either LD or Pontiac fever does not occur. Laboratory transmission from bacterial cultures to humans has not been documented. The lack of contagiousness and the apparent low infectivity of laboratory cultures for humans support studies showing that the infectiousness of the bacterium is enhanced by intra-amebal growth and by special growth conditions and that a sporelike bacterial form, only produced under special conditions, is highly virulent.50,51,52 LD occurs in both sporadic and epidemic form. About 65% to 95% of reported cases are not associated with known epidemics of the disease.30,188 Underreporting of the disease is common because many sporadic cases are treated empirically without diagnostic studies being performed, because of false-negative diagnostic tests, because of underreporting of diagnosed cases, and because only passive surveillance systems are in place to detect disease occurrence. Thus, only approximately 4200 cases of LD were reported in 2011 (13.5/million population) to the CDC. This rate is lower than that reported in France in 2009, of 18.3 cases/million population, and higher than that reported for England and Wales in 2011, 4.2/million (50% acquired abroad). In contrast, prospective studies of both sporadic community-acquired and nosocomial LD have reported far more cases in the United States than would be expected on the basis of the number of cases reported to the CDC. One prospective community-based study in Ohio of adult patients with community-acquired pneumonia requiring hospitalization found that 2.4% of such patients had LD and that the disease incidence was approximately 80/million population per year.189 When extrapolated to the entire population of the United States, the authors estimated that between 8000 and 18,000 cases of LD occur annually among adults requiring hospitalization for pneumonia. The incidence of LD causing community-acquired pneumonia not requiring hospitalization is not known. One small regional U.S. study estimated that the incidence of LD among outpatients treated for pneumonia was 40 to 280/million population/year.190 Thus, somewhere between 18,000 and 88,000 cases of LD are estimated to occur per year in the United States, the majority of which are neither epidemic nor hospitalized. A German study of community-acquired LD that used sensitive diagnostic tests found that the annual rate was 180 to 360 cases/million population, which, if extrapolated to the United States, would mean that 56,000 to 113,000 LD cases occur per year in the United States191 and that only approximately 2% to 5% of U.S. cases are currently reported to the CDC. Some geographic regions appear to have more LD than others, such as western Pennsylvania and Ohio in the United States and Catalonia in Spain; whether this is due to true differences in disease incidence or better case ascertainment is uncertain. The incidence of LD in the United States and elsewhere appears to be increasing on the basis of the numbers of cases reported to public health agencies; whether this is due to more widespread use of the urine antigen test, better reporting and surveillance, or to a true disease increase is unknown.192 Estimates of LD as a cause of community-acquired pneumonia requiring hospitalization in adults range from 0.5% to 10% of all admitted pneumonia cases; an average value is probably approximately 2%, even in geographic regions with excellent diagnostic capabilities.193–197 LD of children is thought to be uncommon, representing 1% or less of causes of pneumonia in this age group, and generally occurs as a nosocomial disease of immunosuppressed children.198 Neonates may be at relatively high risk of LD because of the immaturity of their immune systems. Both nosocomially and domestically acquired cases of the disease have been reported in apparently immunologically normal newborns exposed to Legionella-contaminated water in incubators, bath tubs, and during birth.198–200 Shortly after the 1976 Philadelphia outbreak, nosocomial LD outbreaks were reported in several cities throughout the United States and Europe. Because relatively little was known about the environmental ecology of Lp, or about optimal diagnostic methods, these outbreaks were characterized by long durations, often years in length, and high numbers of cases and fatalities. For example, the LD outbreak at the Wadsworth Veterans Administration Hospital in Los Angeles, California, resulted in more than 250 cases of disease in both patients and visitors over an 8-year period.16,23,201 A 17-year-long outbreak of unrecognized nosocomial LD occurred at another U.S. hospital, involving many fewer patients.202 Nosocomial LD epidemics continue to occur worldwide, albeit with durations measured in weeks rather than years.203,204 Nosocomial pneumonia usually only affects a relatively small number of hospitalized patients, with attack rates less than 1% of patients.16,205 During nosocomial outbreaks of LD, the minority (5% to 11%) of patients with nosocomial pneumonia of all etiologies have been reported to have LD,16,206–208 although in some explosive outbreaks involving a single ward the attack rates may be much higher. Community-based LD epidemics continue to occur more than 36 years after the 1976 Philadelphia outbreak, some of them quite extensive. Some recent large outbreaks have been in Murcia, Spain, in 2001, involving up to 700 people, with 6 deaths; Barrow-in-Furness, England, in 2002, involving more than 130 people with six deaths; and Quebec City, Canada, in 2012, involving 180 people with 13 deaths.209 Of note, several recent outbreaks involved people visiting a town center, rather than with being inside a certain building. Long-distance spread of disease from industrial aerosols has been reported from France and Norway.25,210 LD affecting travelers constitutes up to half of reported cases in some countries. In many cases, a common source outbreak has been found, but for others the cases appear to be sporadic. Multiple common source outbreaks have been uncovered in travelers by a cooperative European reporting system that collects and analyzes LD cases in travelers.211 Such a well-coordinated traveler’s health monitoring and analysis system is not as well established in the United States, making detection of small travel-associated outbreaks of the disease less likely.212,213 Despite the immense publicity usually generated by epidemics of LD, sporadic cases of the disease are about fourfold more common than linked cases of the disease. Some sporadic cases are undoubtedly the result of common source exposures. This is especially true of travelers who return to their homes during the incubation period, or while they are ill but still well enough to travel. Evidence for common source links for apparently unrelated cases comes from observations that proximity of residence to wet cooling towers is a risk factor for LD.214,215 In one study, LD risk was approximately 1.5 times greater for those living within 1 km of a cooling tower than for those living more than 6 km from a tower; the authors estimated that approximately 20% of sporadic LD could be attributed to community cooling tower exposure.215 Residential acquisition from drinking and bathing water, which rarely results in more than a single case, accounts for less than 10% to 15% of sporadic LD.216,217,218 Pontiac fever often causes explosive outbreaks of disease with high attack rates. Attack rates of 70% to 90% have been reported from several epidemics.7,185,219–221 LD mortality rates are highly variable, ranging from less than 1% to as high as 80%, depending on the underlying health of the patient, promptness of specific therapy, and whether the disease is sporadic, nosocomial, or part of a large outbreak.30,222–224,225 The lowest mortality rates, around 1%, have been observed in recent large outbreaks of the disease, whereas the highest mortality rates have been reported in untreated nosocomial disease in patients with severe underlying diseases.226 The average fatality rates for sporadic disease are estimated to be approximately 10% to 15%. Fatality rates of nosocomial disease have declined by more than 50% in the United States over the past 20 years; a similar but less dramatic decrease in death rates of community-acquired cases has also been observed.30 The decline in mortality rates appears to be due to better and faster disease recognition, especially through use of the urine antigen test, and more widespread use of empiric therapy for pneumonia that includes drugs active against Lp.30,226 Because of the ubiquity of Legionella spp. in our natural and manmade environments, most people encounter these bacteria frequently. Yet most of those exposed do not get the disease. This is attributed to largely unknown host immune mechanisms and to the lack of sufficient concentrations of virulent Legionella spp. bacteria in small aerosol particles. Even during large outbreaks of LD attributed to cooling tower aerosols, the minority of people in the risk area become ill. The known host risk factors for LD are those that result in decreased local or systemic cellular immunity and those activities that increase the chances of exposure to an infectious aerosol or microaspiration of contaminated water. Also important are environmental and bacterial factors such as relative bacterial virulence, bacterial aerosol stability, organism growth conditions, and factors that facilitate the spread of the bacterium from contaminated water to the host, such as wind direction, relative humidity, and aerosol formation. Male gender, cigarette smoking, chronic heart or lung disease, diabetes, end-stage renal failure, organ transplantation, immunosuppression, some forms of cancer, and age older than 50 years have all been found to be host risk factors for LD.205,217,227,228 The approximately twofold greater risk with male gender may be due to the greater prevalence of cigarette smoking and its complications in males. Cigarette smoking increases risk by approximately twofold to sevenfold. Nicotine directly impairs macrophage restriction of Lp growth, and nicotinic acid, which can be formed from oxidized nicotine, as well as being endogenous, promotes Lp virulence.229,230 Suppression of local or systemic cellular immunity, and particularly glucocorticoid administration, increases the risk by approximately twofold to sixfold; cytotoxic chemotherapy was a risk factor in one study. Anti–TNF-α therapy has emerged as a significant risk factor for LD, including TNF-α antibodies, soluble TNF-α, thalidomide, and lenalidomide.231,232 Anti-CD52 therapy can result in severe LD.233 Lung, but not gastrointestinal tract, cancer is an LD risk factor.227 A variety of hematologic malignancies have also been shown to be important risk factors, especially hairy cell leukemia.206,227 Small single studies of genetic predisposition to LD have shown that polymorphisms in TLR4, TLR5, and mannose-binding lectin produce minor predispositions to the disease65,72,74; it is unlikely that these polymorphisms account for the majority of human risk for LD, and confirmatory studies are necessary to better define the risks. Animal studies implicate both TLR2 and TLR9 as being important in host immunity, but to date no human data exist on the LD associated with polymorphisms in these TLRs.66,67 Recent surgery has been an important risk factor for nosocomial disease, probably because of general anesthetic-caused defects in local lung defense, because Legionella-contaminated water is introduced into the respiratory tract in the perioperative or postoperative periods, or both factors combined.234,235 Alcoholism has been a predisposing condition in only some studies and has never been shown to be a risk factor in multivariate analyses.205,206,217,236 Host risk factors for non-Lp LD appear to be similar to those for Lp infection, although data are limited; most patients with these infections are immunosuppressed or have cancer, with the exception of LD due to Llb.237,238 There appear to be no predisposing host factors for Pontiac fever. Activities that increase the chances of exposure to Legionella bacteria in water increase the risks of disease acquisition. Recent overnight travel, use of well water in the home, recent plumbing work inside the home, disruptions of water supply resulting in “brown” water at the water tap, living in a water distribution network with older plumbing, and using an electric water heater all increase the acquisition risk of community-acquired LD.215,217,218,239,240 Other community, recreational, or travel-related activities increasing the chances of acquiring the disease include the use of, or proximity to, whirlpool spas or hot water spring spas; living in close proximity to a cooling tower; or being near decorative fountains.214,215,228,241,242 Rarely reported risks are near-drowning including during tsunamis10,43,242,243 and delivery by water birth.244 Exposure risk factors for LD due to Llb are much different than for Lp infection, with handling of potting soil, being near dripping irrigated hanging plants, cigarette smoking, and not washing hands after use of the soil being the major risk factors.245 A wide range of nosocomial exposures can result in LD. Almost all involve delivery of Legionella-contaminated water into the respiratory tract and include the use of tap water filled or rinsed: nebulizers, humidifiers, oxygen humidifiers, ventilator tubing, and nasogastric feedings or lavages.246–249 Proximity to a contaminated decorative fountain has transmitted nosocomial LD in several outbreaks.26,250 LD acquired during dental procedures, a rare event, is the result of inhalation of dental drill cooling water aerosols.251 Consumption of Legionella-contaminated ice can also be a risk factor for nosocomial LD.252 These risk factor are in addition to exposure to Legionella-contaminated air originating from a cooling tower.253–255 Rare cases of nosocomial Legionella wound infection have resulted from irrigation or bathing of wounds in Legionella-contaminated water.256,257 Nosocomial transmission has occurred in hospitals, nursing homes, assisted living facilities, a dentist office, and other types of health care facilities. LD is transmitted from the environment to humans by inhalation of an infectious aerosol.258 In an unknown fraction of cases, microaspiration of contaminated water into the lungs is the mode of transmission.249,259 Finally, massive aspiration of contaminated water into the lungs during near-drownings is an unusual but reported mode of transmission of the disease.10,43,242,243 Multiple examples of exclusive aerosol transmission of LD exist, especially in epidemics having a cooling tower, water spa, water fountain, or water mister as the source of disease.1,241,254,255,260–265 In these cases, proximity to the aerosol generator, duration of exposure, and presence in an area downstream of the contaminated device have all been found to be risk factors for disease acquisition. Of note, when reported, either consumption of water at the epidemic site has not occurred in the majority of disease victims, or the drinking water of the outbreak site has been culture negative for Lp. The data supporting microaspiration of water as a major mode of transmission are less convincing, but in some specific reports the evidence is compelling. These data include examples of nosocomial disease in patients whose major risk factor was nasogastric tube irrigation with tap water and interruption of nosocomial outbreaks by substituting sterile water for tap water for drinking and nasogastric tube irrigation.249,259,266,267 Whether microaspiration is the major transmission mode for nosocomial disease is controversial and unproven.268 Experimental animal data are unhelpful in this regard because both aerosol delivery and tracheal instillation produce disease.269,270 Rare reports of peritonitis or bowel abscesses caused by Lp have led to speculation that oral ingestion may be a mode of disease transmission.271–273 In all these cases, Lp pneumonia or empyema occurred concurrently, making unclear which organs were the ones primarily infected. In contrast to the relative ease of producing lethal guinea pig pneumonia by the aerosol or intratracheal route, huge amounts of Lp delivered by oral gavage are cleared rapidly without causing severe pneumonia,274 although another experimental study gave somewhat different results.275 It is unlikely that oral ingestion of Legionella-contaminated water is more than a minor mode of transmission of the disease. Prompt notification of public health authorities of any strongly suspected or confirmed case of LD is critically important for detecting epidemics of the disease and is legally required in many regions. What appears to be only a single case of the disease may be part of an epidemic or the index case. Alert physicians have sometimes detected pneumonia clusters that led to discovery of both emerging and long-standing epidemics of LD.1,241 In addition to reporting nosocomial LD to public health authorities, medical institutions should embark on extensive investigation of even a single case of nosocomial LD.276 This is because more cases may have occurred previously or will appear subsequently. Retrospective review of nosocomial pneumonia cases for a 3- to 6-month period may yield more cases. Prospective laboratory testing of all patients with nosocomial pneumonia for LD can also be useful in finding more cases, for a 3- to 6-month period. Hospital physicians should be notified to consider LD when making diagnostic and therapeutic decisions in patients with nosocomial pneumonia until it is clear that any ongoing nosocomial outbreak has been eradicated. Investigation of both community-acquired and nosocomial outbreaks of LD requires a thorough epidemiologic investigation. The epidemiologic investigation will help generate hypotheses concerning the source of the outbreak and demonstrate risk factors using controlled studies. Environmental testing without concurrent epidemiologic investigation can lead to misleading findings, even when molecular fingerprinting is used to compare clinical and environmental isolates.277,278 It is crucially important to have only a qualified laboratory perform environmental studies because not all environmental microbiology companies are competent in the proper collection, processing, and culturing of water specimens for Legionella spp. bacteria; certification is required in some countries, with a voluntary certification program available in the United States.279 It is crucially important to obtain Legionella bacteria clinical isolates as part of an epidemiologic investigation; this allows molecular fingerprinting and comparison of clinical and environmental isolates. Environmental sampling protocols for outbreak investigation are available.279–281 Rough clues to the environmental source of an LD epidemic can be found in the pace of the outbreak and its geographic distribution. Explosive outbreaks involving tens to hundreds of people over a several-day period are most often due to a contaminated massive aerosol generator, usually a cooling tower, but sometimes a whirlpool spa or misting device. Potable water associated outbreaks may produce as many cases, but generally over a much longer period of time, such as many weeks or months. Potable water–related outbreaks are confined to a single building or building complex with common plumbing. Cooling tower–related epidemics often affect both building visitors and those within several hundred meters up to a 10-km distance from the tower.25 Interior aerosol-generating devices, such as recreational spas and misters, cause disease only in building visitors. All aerosol-generating sources implicated or highly suspected of being a source of epidemic LD should be taken out of operation as soon as possible; this generally includes cooling towers, hot tubs, and other such devices. Implicated potable water sources such as plumbing systems should be shut down if possible. A number of guidelines exist for emergency disinfection of such sources and may differ according to local regulations or guidelines.276,281–283 Long-term remediation can be complex and requires expert engineering and public health advice. LD causes acute consolidating pneumonia that cannot be accurately differentiated on initial presentation from pneumococcal pneumonia. Several prospective studies have shown that the two diseases have nearly identical clinical and roentgenographic findings and that nonspecific laboratory test results cannot differentiate between the two diseases.284–286 However, initial clinical findings of both community-acquired and nosocomial epidemic LD seemed to show that a distinct clinical syndrome was observed.287 This syndrome was characterized by fever with pulse-temperature dissociation, myalgia, nonproductive cough, few pulmonary symptoms, diarrhea, confusion, hyponatremia, hypophosphatemia, and elevated liver-associated enzymes. Although this symptom complex does occur in LD, it is neither specific nor frequent enough to allow differentiation of LD from other common causes of community-acquired pneumonia. Clinical scoring systems to help increase diagnostic accuracy have not performed well enough to use to guide proper therapy.288–290 The clinical findings of non-Lp LD do not differ significantly from those caused by Lp.238,291 Llb LD occurs predominantly in nonimmunosuppressed patients, whereas the reverse is true for LD caused by the other non-Lp Legionella spp. Those with immunosuppression and non-Lp LD tend to have a more severe course and greater likelihood of nonpulmonary LD, but this is similar to the findings of immunosuppressed patients with Lp LD. A prodromal illness may occur, lasting for hours to days, with symptoms of headache, myalgia, asthenia, and anorexia. Fever accompanies this prodrome, except in severely immunocompromised patients. Multiple rigors may occur, as well as diarrhea and abdominal pain. Cough, with or without chest pain, may develop hours to days after onset of the prodrome; the cough produces purulent sputum in only about half of patients. The initial clinical picture may be confusing because the systemic symptoms can be more impressive than ones referable to the lower respiratory tract, leading some physicians to diagnose “influenza,” a gastrointestinal illness, and in some cases an acute abdomen syndrome. These prodromal symptoms escalate in severity as the disease progresses, which may result in a several-day delay before presentation; the median time to presentation after onset of illness is around 4 days.292 Careful physical examination of the chest and chest roentgenography almost always demonstrate findings of pneumonia, including focal rales and alveolar filling pulmonary infiltrates that vary from patchy infiltrates to multiple areas of consolidation.293,294 Chest computed tomography findings include ground-glass opacities and consolidation.295,296 Pleuritic chest pain, sometimes in concert with hemoptysis, may occur and can result in an incorrect diagnosis of pulmonary infarction. Headache, so severe as to suggest subarachnoid hemorrhage, may be the most prominent feature. Mental confusion is commonly reported in some series; obtundation, seizures, and focal neurologic findings occur less frequently.297,298,299 On presentation, some patients may have negative chest x-ray films that show focal or diffuse pulmonary infiltrates a day later. Lung cavitation is seen almost exclusively in immunosuppressed patients.300 Pleural effusion without pulmonary infiltrates may rarely be observed as the sole radiographic abnormality. Bronchoscopic findings in patients with consolidating pneumonia are often remarkable for the absence of inflammation or purulent secretions in large airways. Abdominal examination may reveal generalized or local tenderness and, in rare cases, evidence of peritonitis. Splenomegaly is uncommon. Findings of pericarditis, myocarditis, and focal abscesses are rare. No rash is associated with this disease, except that caused by other factors such as drug therapy. Symptoms of rhinorrhea, chronic afebrile fatigue, and fever without pneumonia lasting for many weeks are either not seen or so rare as to make the diagnosis unlikely. A number of nonspecific laboratory test abnormalities may occur in LD.301 These include hyponatremia, hypophosphatemia, increased liver-associated enzymes (aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase), hyperbilirubinemia, leukopenia, thrombocytopenia, disseminated intravascular coagulation, leukocytosis, pyuria, elevated creatine kinase (MM fraction), and elevated lactate dehydrogenase. Some studies have shown that patients with LD are more likely to have hyponatremia than do those with other causes of pneumonia, but the range of serum sodium values is too broad for this abnormality to be diagnostic in an individual patient.302 Laboratory markers of pancreatitis are detected if this is a complication of LD. Renal disease caused by LD may result in the presence of urine casts and white cells, elevated serum creatinine levels, or both types of abnormalities. Myoglobinuria is a relatively common finding and is indicated by a positive dipstick test for “blood” in the absence of significant numbers of red cells in the urine. Hypoxemia occurs in proportion to the severity of the pneumonia and underlying cardiopulmonary disease. Clinical diagnosis may be more specific if the patient’s clinical course after treatment has been given is taken into account and if epidemiologic and immunologic risk factors are considered. The chances of a patient having LD are increased if an acute consolidating pneumonia fails to respond to several days of β-lactam antimicrobial therapy or if the pneumonia is severe enough to require intensive care unit hospitalization. Important epidemiologic clues include use of a hot tub or recreational spa, travel outside the home for 1 or more days, recent pneumonia of a co-worker, fellow conference attendee or fellow traveler, and recent plumbing work done at home or work. Patients with suppression of the cellular immune system are at high risk for getting LD; such patients include those treated with glucocorticoids and with antirejection drugs after organ transplantation. Administration of high-dosage methylprednisolone or muromonab CD3 for acute organ rejection is an especially high-risk factor for LD, as is TNF-α antagonist therapy. The nonspecific presentation of LD can make clinical diagnosis difficult and mandates empiric therapy for this disease in most patients with community-acquired pneumonia of uncertain etiology. Diagnosis of the index or sporadic case of nosocomial LD is difficult because of the rarity of nosocomial LD in most hospitals and because laboratory testing for nosocomial LD is uncommon in such settings. Extrapulmonary infections are rare and usually occur as metastatic complications of pneumonia in immunocompromised patients. Metastatic infection has been reported almost exclusively in immunocompromised patients, or patients with fatal LD, who may develop abscesses and other infections of the brain, spleen or extrathoracic lymph nodes, and skeletal and myocardial muscles.303–307 Other reported sites of metastatic infection have been the intestines and liver, the kidneys, the peritoneum, the pericardium, vascular shunts and grafts, bone marrow, joints, surgical wounds including prosthetic heart valves and aorta, native heart valves, the perirectal area, and the skin and subcutaneous tissues.271,308–319 Both reported cases of culture-confirmed native valve endocarditis were in immunocompromised patients with LD.319,320 In some of these cases the onset of symptoms of the metastatic infection preceded the recognition of pneumonia by several days, and in other cases the metastatic infection presented days to weeks subsequent to onset of the pneumonia. In some cases, the metastatic infection site was the only evidence of relapse of infection. Three cases of metastatic infection have been reported in apparently previously healthy patients,305,321,322 but otherwise immunosuppression is found in such patients. Local extension of a thoracic empyema into the soft tissues of the chest has been reported after thoracentesis.323 Rare cases of primary infection not preceded or accompanied by pneumonia have also been reported. Some of these appear to be the result of direct inoculation of Legionella-contaminated water into various tissues, usually in hosts with immune compromise. Some infections were introduced by bathing postoperative patients with contaminated tap water, by the use of therapeutic baths, and by inadvertent tap water irrigation of the mediastinum after esophageal perforation. In other cases the route and source of infection are unknown. Culture-proven sites of such infections have been surgical or other wounds, subcutaneous tissues, prosthetic and native joints, prosthetic heart valves, the mediastinum, and the respiratory sinuses.256,257,317,322,324–335 In contrast to the case of native valve endocarditis, almost all patients with prosthetic valve endocarditis were neither immunosuppressed nor had LD.324,325,328,329,336–338 Native and prosthetic joint infections have been reported uniformly in immunocompromised patients with preexisting arthritis, with one exception of a patient who was not immunocompromised but did have underlying arthritis.317,322,333–335,339 With one exception all joint infections occurred in the absence of LD. PCR-positive, but culture-negative, meningoencephalitis has been reported in patients without LD.340
Legionnaires’ Disease and Pontiac Fever
Introduction and History
Etiologic Agent
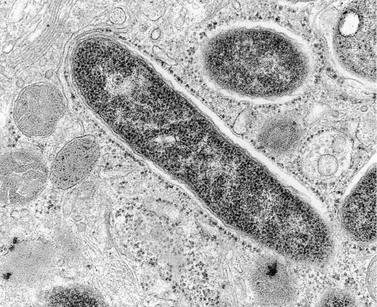
Microbial Ecology
Pathogenesis
Overview
L. pneumophila Virulence Factors
Virulence Factors and Pathogenesis of Other Legionella spp.
Epidemiology
Incubation Period and Contagiousness
Patterns and Rates of Disease and Mortality
Risk Factors
Modes of Transmission
Outbreak Investigation
Environmental Decontamination for Outbreaks
Clinical Manifestations
Legionnaires’ Disease
Extrapulmonary Infections
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Legionnaires’ Disease and Pontiac Fever
234