Laboratory Markers of Endothelial Cell Activation and Dysfunction
Laboratory Markers of Endothelial Cell Activation and Dysfunction
Eduard Shantsila
Gregory Y.H. Lip
Robert P. Hebbel
The diverse functions of the vascular endothelium are of critical importance in maintaining physiologic homeostasis, yet they also are participants in a great variety of disease processes.1,2 These functions include provision of the physical and biologic interface between blood and tissue, integration of disparate signals and coordination of responses, governance of hemostatic balances, regulation of vasomotor tone, management of vascular permeability, participation in immune and inflammatory processes, transduction of mechanosensitivity signals, and operation as an autocrine/paracrine/endocrine signaling network. Yet, the endothelium retains the flexibility to exhibit regional and local variations in these functions as required. Moreover, the endothelial cell lies at the interface of these multiple physiologic functions and thereby fundamentally links them. Illustrative examples of this include the frequent associations of coagulation activation and of vasoregulatory disturbance with inflammatory states.
ENDOTHELIAL CELL ACTIVATION AND DYSFUNCTION
Each of these component biologies exhibits quantitative and qualitative variation (albeit with substantial interdependence), so endothelial phenotype potentially can exist in a complex continuum of states. Nevertheless, the traditional description of the endothelium as being “quiescent,” “activated,” or “dysfunctional” is still conceptually useful in biomedicine. In actuality, however, the normal state of the endothelium is anything but quiescent; rather it contributes active homeostatic adjustments for maintenance of physiologically optimal states, an example being its vigilant husbandry of vasomotor tone. Its normal phenotype is anti-inflammatory, antithrombotic, and antiadhesive.1,2
The term “activated” endothelium generally refers to its state of response—often localized or regional—to a challenge as part of the host response to disease. Typically, such states are adaptive and organized to be of benefit to the host. Examples are the endothelial cell’s participation in the normal inflammatory response, adjustment of hemostatic balance to staunch bleeding or favor fibrinolysis, and its assumption of an angiogenic phenotype as required for wound healing and placentation.1,2 Some activation responses occur rapidly (e.g., shedding of surface adhesion and hemostasis molecules, retraction, decryption of tissue factor [TF], and release of P-selectin, von Willebrand factor [vWF], and monocyte chemoattractant protein-1), while others require gene transcription and protein synthesis (expression of E-selectin, intercellular adhesion molecule [ICAM]-1, vascular cell adhesion molecule [VCAM]-1, plasminogen activator inhibitor [PAI]-1, TF, and platelet activating factor).
In contrast, “endothelial dysfunction” is typically used to describe states in which the extant endothelial biology is harmful to the patient, typically by developing phenotypes with features that are the converse of the normal state. Examples include the counterproductive roles of a dysfunctional endothelium in atherosclerosis,3 chronic renal failure,4 reperfusion injury physiology,5 sickle cell anemia,6 gestational disorders,7 diabetes,8 and sepsis.9 Notably, these all involve widespread, not just localized, endothelial dysfunction, which is pathologic and has escaped from normal, delicate, adaptive control. Not coincidentally, each of these disorders includes disturbance of hemostatic systems and elevated risk for untoward clinical events, particularly thrombosis,10 because an endothelial dysfunctional state potentially perturbs all arms of Virchow triad.
The difficulty arises in attempting to distinguish between activation and dysfunction. From the perspective of the cardiovascular literature, a functional deficiency of endothelial nitric oxide synthase (eNOS) is the sine qua non of endothelial dysfunction, as documented by experimental and physiologic studies.11 Indeed, the eNOS knockout mouse provides the paradigmatic experimental model of endothelial dysfunction and demonstrates that this leads to deficient endothelial-dependent vasodilation, hypertension, increased thrombosis, atherosclerosis, and stroke.12 Accompanying defects include loss of the nitric oxide (NO) inhibitory effect upon platelet aggregation, leukocyte adhesion, smooth muscle cell proliferation, and expression of some surface molecules (e.g., VCAM-1 and TF). In human biomedicine, however, eNOS is not absent but rather becomes functionally inadequate and uncoupled so that it produces superoxide. This state is promoted by disparate conditions, for example, inflammation, oxidative stress, substrate/cofactor deficiencies, elevated levels of the competitive inhibitor asymmetric dimethylarginine (ADMA), oxidized lipids, aberrant wall shear stress, perturbation by microparticles (MP), and others.
However, this eNOS-based definition of endothelial dysfunction is perhaps too restrictive, as the majority of clinical information about it is derived from atherosclerosis and its risk factors and comorbidities, in which deficient NO bioavailability is paramount. Yet, there are various dysfunctional states where it perhaps need not be.10 Endothelial dysfunction additionally involves various mixes of multiple activation-related imbalances leading to a proinflammatory and prothrombotic phenotype, permeability barrier dysfunction, and oxidative stress in addition to disturbed vasoregulation. While inflammation in general, with its attendant oxidant generation, is a major cause of eNOS dysfunction,13 it remains possible that defective endothelial-dependent vasodilation may not be a universal feature of endothelial dysfunction. Indeed, from the hemostasis perspective, endothelial dysfunction is typically regarded to be the provisional interpretation if the patient is put at increased risk for clinical harm.10
Regardless of the definition, these changes provide the basis for the available laboratory testing of endothelial status. Interpretation of these is complicated by the continuum of activation states, and the ability of biomarkers to distinguish between activation and dysfunction is by no means unambiguous. This chapter discusses the four types of available laboratory testing that have sufficient endothelial specificity so as to be useful. Physiologic testing provides the gold standard of evidence for endothelial dysfunction from the cardiovascular perspective. Blood biomarkers and their associations with disease have received the most attention. Circulating MP, although still emerging as a tool, perhaps offers the most potential promise for revealing pathobiologically useful information about the endothelium in situ. Circulating endothelial cells (CECs) are still in development as a potentially useful tool. Cells popularly referred to as “endothelial progenitor cells” (EPCs) cannot be recommended as a biomarker of endothelial dysfunction. Predictably, most clinical information on validity and utility of these biomarkers is derived from the atherosclerosis context. However, that is advantageous since there is a wealth of information regarding all aspects of atherosclerosis, in particular its state of clinically inapparent, subtle inflammatory involvement that is tightly associated with endothelial dysfunction.
PHYSIOLOGIC ASSESSMENT OF ENDOTHELIAL FUNCTION
Dynamic regulation of vascular tone by the endothelium is realized by its production of substances having either vasoconstrictive or vasodilatory properties, the balance of which (along with other inputs) governs vascular hemodynamics. The major endothelial-derived vasodilator is NO,14,15 which additionally contributes to several other aspects of vascular health, as noted above. In aggregate, these exert antiatherogenic effects.16 Because endothelial dysfunction typically involves impaired endothelialdependent vasodilatation, its investigation can provide useful information even if clinical focus is not upon vasoregulation per se. Indeed, physiologic assessment is the only method that directly identifies endothelial dysfunction.
Invasive Assessment
Angiography—the Gold Standard
Direct angiographic measurement of the change of coronary artery diameter, blood flow, and vascular resistance in response to an intracoronary infusion of acetylcholine (ACH) is considered to be the gold standard in endothelial function assessment.17,18 Normal epicardial and microvascular coronary arteries respond to this by an increase in diameter and blood flow. In subjects with endothelial dysfunction, ACH causes paradoxical vasoconstriction and a decrease in coronary blood flow due to the disruption of NO-dependent vasodilation.
Plethysmography. Venous occlusion plethysmography records changes in the forearm volume after occlusion of blood circulation to the hand. Thereby, changes in vascular resistance after intravascular administration of vasoactive drugs can be assessed as a measure of endothelial function. The method is accurate and reproducible, but it is semi-invasive and labor intensive, thus limiting its clinical utility.19,20
Noninvasive Assessment
Imaging
Noninvasive assessment of coronary artery endothelial function can be performed using positron emission tomography imaging or contrast ultrasound by measuring the change in myocardial blood flow in response to pharmacologic (e.g., adenosine, dipyridamole) and/or physiologic (cold pressor test) stimuli. This technique permits calculation of coronary flow reserve as a parameter reflecting vascular smooth muscle and endothelial contributions to microcirculatory vascular function.21
Peripheral Arteries
Several methods have been developed for the assessment of peripheral artery endothelial function as a surrogate marker of coronary arterial endothelial dysfunction. Indeed, measurement of the vascular response to reactive hyperemia after arterial compression has become the most popular approach for the assessment of it. Flow-mediated dilation (FMD) tests conduit artery function, while peripheral artery tonometry (PAT) tests microvascular function.
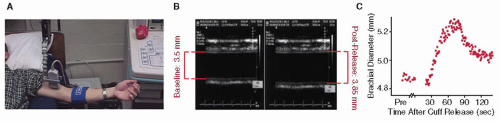
The FMD response is recorded by brachial artery ultrasound imaging (FIGURE 48.1). This comprises the current noninvasive gold standard method, one that is extensively validated, has a solid biologic foundation, and correlates with coronary endothelial function.22,23 Yet, it is very operator-dependent, exhibits diurnal variation, and requires substantial experience in order to achieve reproducible results.24 Nonetheless, it is the evaluative tool that has been most widely deployed and for which the most data exists. PAT, on the other hand, is virtually observer-independent and is performed using a system that calculates a reactive hyperemia index based on the contralateral finger serving as control. The device has been approved by the U.S. FDA and has been validated in different populations. PAT also correlates with coronary endothelial dysfunction and FMD, and assessment of vascular health using it has been demonstrated to be feasible in outpatient settings.25,26 Therefore, it is emerging as a potential clinical tool to identify patients at high risk for future cardiovascular events (CVE).26 The relative limitation of the latter approach is the high cost of consumables.
Controls
It is essential that these approaches be accompanied by similar evaluation of endothelial-independent vasodilation (e.g., in response to a NO donating drug) so that the meaning of observations is not misinterpreted.
Clinical Utility of Vasoregulatory Assessment
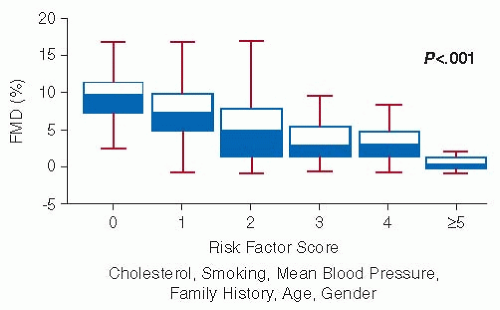
Endothelial dysfunction is understood to be the earliest detectable vascular abnormality in the natural history of atherosclerosis and atherothrombosis and, therefore, is believed to have predictive value.27 It is closely associated with the presence of traditional cardiovascular risk factors.28 For example, even in the absence of known atherosclerosis abnormal FMD is observed in smoking, hypercholesterolemia, obesity, diabetes, and hypertension, and FMD deterioration parallels accumulation of such risk factors (FIGURE 48.2). This has been interpreted as supporting the concept that the endothelium serves as the ultimate integrator of all cardiovascular risk inputs (environmental, genetic, unknown) and, thus, provides a meaningful way to identify overall risk even in patients not having classical atherosclerotic risk factors.18,29
FIGURE 48.1 Measurement of FMD at the brachial artery. A: Placement of the ultrasound probe proximate to the site of transient arterial occlusion. (Kindly provided by Dr. Aaron Kelly, Pediatrics, University of Minnesota). B: Signal output of arterial wall diameter during rest (left) and postocclusion hyperemia (right). (Reproduced from Barac A, et al. Methods for evaluating endothelial function in humans. Hypertension 2007;49:748-760, with permission from Wolters Kluwer Health.) C: Representative results from the multiple measures over time for a given patient. (Reproduced from Corretti A, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery; a report of the International Brachial Artery reactivity task force. J Am Coll Cardiol, 2002;39:257-265, with permission from Elsevier.)
Endothelial dysfunction identified by aberrancy of endothelial-dependent vasodilation predicts future disease progression and is independently associated with future CVE.18,29,30 In established coronary artery disease (CAD), it can predict future acute vascular events.18,29 The major extant issue regarding the clinical applicability of FMD is that virtually all data are from cohort studies, such that applicability to the individual patient has not been robustly and prospectively studied. Yet, lending support to the belief that it meaningfully reflects biologic endothelial dysfunction is the improvement of FMD by therapeutic interventions that improve cardiovascular prognosis32 such as statins,18 weight loss,33 smoking cessation,34 and control of hyperglycemia35 and hypertension.36
FIGURE 48.2 Inverse relationship between FMD and accumulating coronary risk factors in asymptomatic adults. Presence of hypercholesterolemia, smoking, hypertension, family history, age, and gender are represented by a proportionate, composite risk score. (Adapted from Celermajer DS, et al. Endothelium-dependent dilation in the systemic arteries of asymptomatic subjects relates to coronary risk factors and their interaction. J Am Coll Cardiol 1994;24(6): 1468-1478, with permission from Elsevier.)
Aside from atherosclerotic disease, abnormal FMD was observed in patients who had previously developed idiopathic venous thrombosis,37 as well as in those with antiphospholipid antibody syndrome.38 In patients with systemic lupus erythematosus, FMD has been observed to be39 and to not be40 abnormal, although blood biomarkers of endothelial dysfunction do tend to be abnormal (see below).
Caveats
Evaluation of FMD has been performed mostly in chronic conditions in which endothelial dysfunction would be strongly expected given the clinical context. Its potential value in many nonatherosclerotic disease settings, for example, sepsis, is a largely unstudied possibility, and interpretative caution is necessary, as FMD can be transiently diminished by unrelated concurrent situations (e.g., an acute viral illness).31 Abnormal FMD has interestingly been reported in some unexpected situations, for example, migraineurs,41 Alzheimer disease,42 children of hypertensive parents,43 rheumatoid arthritis,44 and sarcoidosis.45 It is reported to be alleviated by aromatherapy for night-shift workers.46 It is not yet clear whether such observations reflect the substantial issues regarding test performance47 or its specificity or the nascence of biomedical understanding of some pathophysiologies. The major source of variability in FMD measurement is between subjects, and the normal range is sufficiently large that definitive identification of endothelial dysfunction in an individual patient is difficult.
SOLUBLE MARKERS OF ENDOTHELIAL DYSFUNCTION
Direct, specific, and quantifiable detection of endothelial surface molecule expression on the activated/dysfunctional endothelium in vivo in clinical settings eventually will likely be achieved through engineering advances in reporter nanoparticles and imaging technologies. In lieu of such definitive testing, states of endothelial activation or dysfunction currently are often inferred from the increased levels of the soluble forms of these endothelial-derived molecules in the blood.
General Cautions
Substantial caveats apply to examination of circulating biomarkers. Of those used to probe endothelial cell function, only E-selectin is endothelial-specific; all others can be derived from other cellular sources and, therefore, reflect the biology of those as well. The usual reported levels of blood biomarkers derived from endothelial cells are based on plasma samples that do not distinguish between their truly soluble versus MP-bound forms. Levels of biomarkers in human disease overlap with their normal ranges, so data on their clinical instructiveness often have been derived from comparison of ends of the value spectrum (e.g., lowest versus highest quintiles). Additionally, virtually all studies rely upon single measurement at any given point of time, so that they are affected by normal variability, including diurnal variation for some. Although patients with documented endothelial dysfunction (by FMD) tend to have blood elevations of endothelial-derived biomarkers, there has been no comprehensive examination of the robustness of their independent association with documented endothelial dysfunction.
In general, these issues render biomarker measurements more useful for large patient studies than for application to the individual patient. Moreover, some biomarkers appear to retain sufficient functional characteristics so as to actively influence disease pathobiology. Thus, clinical correlates with biomarker elevations potentially can be different in different disease contexts. Nonetheless, they have been widely used, and some do have predictive value.
Endothelial Adhesion Molecules
The vascular endothelium exhibits carefully orchestrated modulation of its surface to govern its coordinated interactions with circulating blood cells. This involves multiple adhesion molecules, cytokines/chemokines and other mediators.48 The principal endothelial molecules are P- and E-selectins that mediate initial tethering/capture/rolling, and VCAM-1/ICAM-1 that enforce stronger and stationary adhesion. Experimental studies have revealed that increased endothelial expression of such molecules is a typical and participatory feature in the development of atherosclerosis.49,50,51,52 For example, knockout of either E- or P-selectin renders mice less susceptible to atherosclerosis, and knockout of both has additive benefits.53,54
Of note, however, blood levels of endothelial adhesion molecules do not necessarily quantitatively reflect the phenotype of the endothelial surface in situ because the levels of these soluble forms mostly reflect activation of the differing mechanisms causing their specific shedding (i.e., a cleavage event, shedding of MP, or release of an alternatively spliced variant). Thus, biomarker elevations in blood can occur in parallel with reciprocal diminished expression on the cell surface, and sometimes are seen even in the context of their concurrently diminished transcription. Due to such regulated shedding, their soluble forms (sP-selectin, sE-selectin, sVCAM-1, sICAM-1) are typically—but variably— increased in states of endothelial activation and dysfunction, and they have often been used to presumptively identify such states.
So, within the data set from a study focused on a specific disease context, a higher level of soluble endothelial adhesion molecules can be associated with worse endothelial dysfunction (as documented by FMD),55,56 although this discrimination can require the within-study empirical identification of a high/low cutoff point. Conversely, soluble adhesion molecules can be elevated in active vascular disease absent abnormal FMD, as reported for lupus patients,57 emphasizing that such elevations can reflect endothelial activation without actual (apparent) dysfunction, as narrowly defined by abnormal FMD. The resulting levels of these molecules tend to track together,58 so sorting out their independent predictive value for future trouble even in the context of endothelial dysfunction can only be done via large studies (Table 48.1).
Endothelial Selectins
E-selectin has the greatest degree of endothelial specificity, it is not constitutively expressed normally, and it becomes expressed on the endothelial surface upon activation in the inflamed endothelium.50 Yet, sE-selectin has been disappointing as a biomarker for prediction of future CAD in prospectively evaluated healthy subjects.58 It may or may not be useful to indicate injury responses or cardiac disturbances.59,60,61 Of interest, sE-selectin was observed to be elevated in lupus vasculopathy patients having thrombotic risk.62
P-selectin is stored in Weibel-Palade bodies and is constitutively expressed on endothelial cells at extremely low levels. It is also expressed in megakaryocytes and is stored in the alpha granules of platelets. Its surface expression on the endothelium and appearance in the blood (perhaps due to both cleaved and alternatively spliced forms63) can robustly increase in response to varied stimuli,50 so plasma sP-selectin often parallels endothelial activation/dysfunction. However, utility vis-à-vis endothelial status is negated by the nonspecificity of its origin (i.e., from platelets also). Elevated sP-selectin can add confirmatory information in acute thrombosis and has predictive value in terms of recurrent deep vein thrombosis and future venous thromboembolism in patients with and without cancer.64 However, a large prospective trial did not support any value for sP-selectin in predicting future CVE in patients with CAD58(Table 48.1).
Endothelial Cell Adhesion Molecules
The elaboration of soluble forms of endothelial CAMs by inflammatory cells confers some ambiguity upon interpretation of their typically elevated levels in inflammatory disease contexts. Endothelial VCAM-1 plays a prominent role in initiation and progression of atherosclerotic lesions.49,50,65,66 Yet elevated sVCAM-1 levels had no predictive value for subsequent development of CAD or events (CVE) in prospective studies of healthy men58,67,68(Table 48.1). On the other hand, acute elevations predictably occur in acute coronary syndromes (ACS) in which higher levels may identify those having increased risk for future adverse CVE.69,70 In type 1 diabetics, higher sVCAM-1 levels were associated with subsequent development of retinopathy, albuminuria, and cardiovascular disease.71
Endothelial ICAM-1 plays a similar pathogenetic role in atherosclerosis and is similarly released by nonendothelial cells in inflammatory states.49,50,51 Yet, elevated sICAM-1 levels do appear to have value for assessing endothelial dysfunction in the atherosclerosis context (Table 48.1), in which they are tightly associated with levels of both C-reactive protein (CRP) and vWF.58 Three large prospective studies of healthy men demonstrated that the highest elevations of sICAM-1 predict future CAD or CVE, independent of CRP or traditional risk factors.58,67,72 Oddly, a large prospective study of healthy women found that while higher sICAM-1 levels predicted CAD progression and even the need for coronary bypass, they did not predict CVE.73 Increased sICAM-1 levels predicted subsequent stroke risk in type 2 diabetes74 but not in healthy white women.75 Thus, the utility of elevated sICAM-1 levels may be context dependent. Interestingly, temporally increasing sICAM-1 levels were associated with all-cause mortality for men and women older than 64 years.76 In systemic lupus erythematosus, sICAM-1 elevations predict future first cardiac events.77
Table 48.1 Predictive value of circulating endothelial-derived adhesion molecules for cardiovascular disorders
In ACS setting, sICAM-1 levels are elevated and parallel the increased expression of the monocyte receptor CD11b78 and higher CRP and titers of antibody to oxidized low-density lipoprotein.79 After an acute MI, higher levels of sICAM-1 measured 7 days or 10 weeks later predicted subsequent coronary symptoms and events.69,80,81 Similarly, for hypertensive type 2 diabetes patients, sICAM-1 level paralleled degree of atherosclerosis.82 However, data are not uniform, as studies following up ACS70 or coronary bypass grafting83 observed no predictive value of sICAM-1 for subsequent CVE.
Perhaps most strongly supporting the value of sICAM-1 measurement, its elevation correlated with impairment of FMD in obese hypertensive children and adolescents,84 and even in apparently healthy adolescents.85 The elevated sICAM-1 level in smokers decreases upon smoking cessation, perhaps bolstering its value as a meaningful marker.86
Thus, taken together these data identify elevated sICAM-1 levels as having considerable potential as a useful prognostic marker for increased risk of CAD and future CVE, but broader application to clinical predictions in endothelial dysfunction generally is not known. So overall, more study of its value in the clinical setting is necessary. The other candidate markers (sVCAM-1, sE- and sP-selectin) can all be elevated in endothelial activation states (with sVCAM-1 being the most commonly used indictor), but they cannot be relied upon to distinguish between activation and dysfunction. Indeed, the usefulness of soluble adhesion molecules, either independently or in various combinations, for predicting or quantifying measured FMD has not been comprehensively studied.
Hemostasis and the Maintenance of Blood Fluidity
Endothelial integrity is fundamental for separation of blood components from thrombogenic subendothelial materials, and the luminal surface of the healthy endothelium itself is anticoagulant and antithrombotic1,2,10 (see Chapter 42). For example, it displays thrombomodulin (TM) and the endothelial protein C receptor (EPCR), a glycocalyx carrying antithrombin (AT) and tissue factor pathway inhibitor (TFPI); it elaborates tissue-type plasminogen activator, along with low levels of PAI-1; and it has an absence of TF. Further, the endothelium produces molecules that impede platelet reactivity, particularly NO. These prominent antithrombotic characteristics can be challenged during endothelial activation and are typically changed dramatically in damaged or dysfunctional endothelium.10 However, few of the consequent blood alterations are sufficiently dominated by endothelial biology so as to offer utility in detecting endothelial status vis-à-vis activation and/or dysfunction.
Thrombomodulin
Although TM is also expressed by monocytes and other nonendothelial cells, its thrombin-stimulated conversion of protein C to activated protein C (APC) is understood to be predominantly endothelial in location, where this is robustly augmented by its companion molecule, EPCR. Inflammatory mediators (particularly tumor necrosis factor α [TNFα]) elevate blood sTM levels but conversely diminish endothelial TM expression, thereby lowering APC generation capability. The latter is observed in a number of inflammatory vascular syndromes associated with endothelial dysfunction, sepsis perhaps being the paradigmatic example,9 but others include vasculopathies, diabetic angiopathy, and acute respiratory distress syndrome.87,88 Yet no direct comparison to measured global endothelial dysfunction (e.g., by FMD) has been reported. Moreover, protein C biology is heavily influenced by endothelial-independent events and activities.
Nevertheless, low APC levels are independently associated with increased risk of CAD and some CVE,89 and deficient conversion of protein C to APC characteristically identifies adverse prognosis in sepsis syndromes and reperfusion injury.90,91 Conversely, increased plasma levels of sTM are associated with increased mortality from trauma and acute respiratory distress syndrome.87 In sepsis patients, in whom APC generation is variably impaired,9,91,92 higher sTM levels presage development of DIC (disseminated intravascular coagulation), multiorgan failure, delayed recovery, and mortality.87,93 For apparently healthy subjects not having CAD, higher sTM levels identified diminished risk for future coronary and cerebrovascular events.94 Yet in those having established CAD, higher sTM identified enhanced risk for future CVE.95
It has consequently been suggested that sTM level possibly has varying implications depending upon the specifics of the extant vascular biology in different clinical contexts. Possibly additionally contributing to biologic heterogeneity, multiple TM cleavage sites underlie sTM release. Some, but not all, of the resulting fragments retain anticoagulation activity.96 The extent to which different TM fragments (retaining varying functional domains) impact differentially upon biology requires exploration. Whether elevated sTM levels can generally assist with the fine distinction between endothelial activation and dysfunction is debatable, yet their association with certain catastrophic endothelial failures is clear.
Von Willebrand Factor
Plasma vWF levels are mostly endothelial in origin, despite some contribution from platelets.97 Release of stored high molecular weight (HMW) forms via Weibel-Palade body exocytosis98,99 is an immediate-early endothelial response prompted by a spectrum of (patho)physiologic biologic mediators. Thereafter, its functional biology is dominated by anchorage to membrane P-selectin100 and cleavage by liver-derived ADAMTS-13 (A Disintegrin And Metalloproteinase with a Thrombospondin type 1 motif)101 to preempt the strong prothrombotic effect of HMW vWF.102 Therefore, the largest forms of vWF are observed in plasma only under certain disease conditions,103,104,105,106 and analysis of the vWF size distribution has not been found to be of value in assessment of endothelial function. On the other hand, high plasma vWF levels are associated with increased prospective risk for arterial thrombosis,107 CAD,108 and stroke109 in the healthy population, and for future CVE in angina patients.110 However, risk defined by vWF tracks closely with that defined by traditional risk factors and Framingham risk calculation, so it adds little value beyond classical risk assessment.108,111 Interestingly, while vWF elevations in the atherosclerosis context are accompanied by inflammatory markers (e.g., sICAM-1, CRP), similar elevations in the apparently healthy that predict venous thromboembolism are not associated with these.112 This may be an important clue vis-à-vis potential utility of vWF assessment more broadly in consideration of thrombotic risk.
Although measurement of plasma vWF has been used very widely to evaluate endothelial function, sometimes as the sole basis for assignment of “endothelial dysfunction,” its utility is perhaps compromised by being too sensitive as a biomarker. Elevation of plasma vWF is probably a universal feature of endothelial dysfunction,113 in part because the associated deficiency of endothelial NO production is the major endogenous inhibitor of Weibel-Palade body exocytosis.98 Experimentally, pathologic vWF release and a consequent thrombotic renal microangiopathy occur in a relatively pure model of endothelial dysfunction, the eNOS knockout mouse.114 Consistent with this illustrative association between vWF release and endothelial dysfunction, vWF elevations independently correlate with deficient brachial artery FMD in ACS,115 hypertension,116 and congestive heart failure117 (FIGURE 48.3). However, elevated vWF levels are equally predictable in the disparate situations involving inflammatory activation since vWF behaves like an acute phase reactant.113,118 It is further noteworthy that vWF levels vary with age, gender, race, and, in particular, genetics.107 Overall, it seems improbable that vWF levels can distinguish between endothelial activation and dysfunction, although this issue certainly deserves more thorough clinical correlation and experimentation.
Only gold members can continue reading. Log In or Register to continue
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient



